Opieka okołooperacyjna
Opioidowe leki przeciwbólowe
dr n. med. Jarosław Woroń
Prawidłowo dobrane i dawkowane leki przeciwbólowe pozwalają na szybszą mobilizację pacjenta, zmniejszają częstość powikłań oraz prawdopodobieństwo przetrwałego bólu pozabiegowego.

dr n. med. Jarosław Woroń
Wstęp
Opioidy są jedną z głównych grup leków stosowanych w terapii bólu. Jak pokazuje doświadczenie, racjonalna farmakoterapia bólu, oparta na dobrej znajomości mechanizmów działania leków, ich farmakokinetyki, działań niepożądanych oraz interakcji z lekami z innych grup pozwala skutecznie łagodzić ból u większości, bo aż u 85-95% pacjentów.
Farmakologiczne mechanizmy działania analgetyków opioidowych
Opioidy działają bezpośrednio na trzy typy receptorów opioidowych: μ (MOR), δ (DOR) oraz κ (KOR). Receptory opioidowe zlokalizowane są w strukturach ośrodkowego i obwodowego układu nerwowego. Należą one do rodziny receptorów sprzężonych z białkami G. W następstwie aktywacji, po przyłączeniu ligandu, receptor opioidowy zmienia swoją konformację, efektem czego jest przyłączenie swoistego białka błonowego związanego z GTP – białka G. Białko G składa się z trzech podjednostek: α, β i γ. Połączenie się białka G z odpowiednią domeną receptora opioidowego powoduje jego fosforylację i rozbicie na podjednostki. Podjednostka α białka G inicjuje dalsze skomplikowane procesy wewnątrzkomórkowe. W przypadku opioidu jego oddziaływanie z receptorem zlokalizowanym w błonie komórkowej powoduje zmianę właściwości związanego z nim białka Gi (pierwszy układ sygnałów) i zahamowanie aktywności cyklazy adenylanowej – enzymu odpowiedzialnego za produkcję cAMP w komórce (drugi układ sygnałów). Powoduje to zamknięcie kanałów wapniowych oraz aktywację transportu jonów potasu do przestrzeni zewnątrzkomórkowej, a w efekcie hiperpolaryzację błony komórkowej. W następstwie tych zmian zostaje wstrzymane uwalnianie pronocyceptywnych neuroprzekaźników oraz zwolnione lub zahamowane przewodzenie impulsów we włóknach nerwowych.
Opioidy stanowią grupę leków, które różnią się powinowactwem do trzech typów receptorów opioidowych (μ, δ, κ), rodzajem interakcji z receptorami opioidowymi (agoniści, częściowi agoniści, antagoniści), właściwościami fizykochemicznymi cząsteczki (wielkość, lipofilność), okresem półtrwania i charakterystyką farmakokinetyczną, a więc przebiegiem procesów wchłaniania, dystrybucji, metabolizmu i wydalania oraz oddziaływaniem na zstępujące układy kontroli bólu (tramadol, tapentadol) i receptory N-metylo-D-asparaginianowe (np. metadon).
Ze względu na siłę działania przeciwbólowego opioidy dzielimy na słabe i silne. W praktyce oznacza to, że słabe opioidy, klasyfikowane na drugim stopniu drabiny analgetycznej, charakteryzują się efektem pułapowym (tzn. powyżej pewnej dawki nie obserwujemy nasilenia działania analgetycznego, a zwiększa się ryzyko działań niepożądanych). Podział ten wydaje się szczególnie użyteczny w codziennej praktyce klinicznej. Dostępnymi w Polsce słabymi lekami opioidowymi są: kodeina, dihydrokodeina i tramadol, a do silnych opioidów stosowanych w naszym kraju należą: morfina, fentanyl, buprenorfina, metadon, tapentadol, oksykodon oraz połączenie oksykodonu z naloksonem.
Parametry farmakokinetyczne analgetyków opioidowych opisano w tabeli 1.
Słabe opioidy
Kodeina
Kodeina jest alkaloidem fenantrenowym, pochodną opium (3-metylomorfina), agonistą receptora opioidowego μ o sile działania 10-krotnie słabszej od morfiny. Jest metabolizowana przy udziale izoenzymu CYP2D6 na drodze O-demetylacji do morfiny oraz na drodze N-demetylacji do norkodeiny, która nie ma istotnego znaczenia klinicznego. Pierwszy szlak metaboliczny i powstająca w jego efekcie morfina odgrywa zasadniczą rolę w działaniu przeciwbólowym kodeiny. Jak pokazują badania, aktywność izoenzymu CYP2D6 jest bardzo zróżnicowana w zależności od populacji. Izoenzymu tego nie ma 6-14% osób rasy białej, 1-2% populacji azjatyckiej oraz 1-18% mieszkańców Afryki w zależności od rejonu, co czyni ich niewrażliwymi na kodeinę (tzw. wolny metabolizm). U kilku procent populacji obserwuje się natomiast jego nadekspresję (tzw. bardzo szybki metabolizm leku), w następstwie czego zachodzi szybsza przemiana kodeiny do morfiny. Chociaż wydaje się, że z uwagi na małe ilości morfiny powstającej z kodeiny ryzyko groźnych powikłań w tej grupie chorych jest niewielkie, nie wolno lekceważyć tego zjawiska.
Kodeina najczęściej jest podawana drogą doustną w postaci kropli lub tabletek o natychmiastowym uwalnianiu. Zaczyna działać po upływie 15-30 minut, a czas jej działania wynosi około 4 godzin. Zazwyczaj jest stosowana w dawce początkowej 20 mg co 4 godziny, maksymalna dawka dobowa wynosi 240 mg. Z uwagi na profil farmakokinetyczno-farmakodynamiczny (trudny do przewidzenia początek działania leku, długi czas latencji efektu analgetycznego) kodeina nie jest dzisiaj zalecana w leczeniu bólu. Wchodzi też w interakcje z wieloma lekami. Jest bezwzględnie przeciwwskazana przed ukończeniem 12 r.ż. z uwagi na ryzyko zgonu.
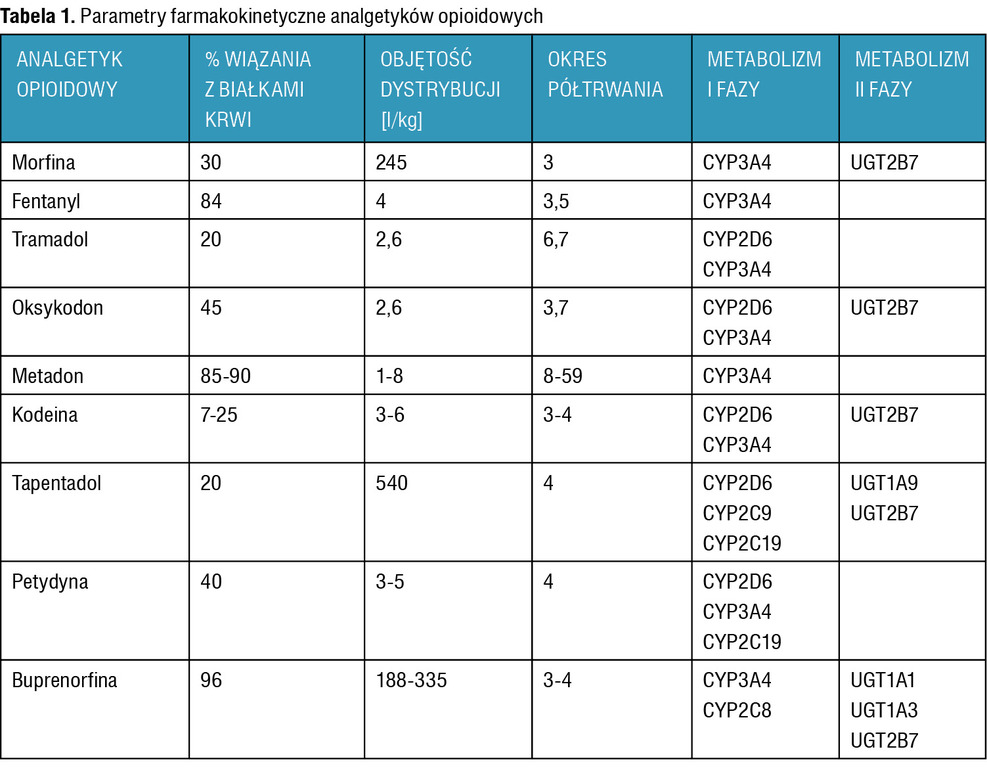
Tabela 1. Parametry farmakokinetyczne analgetyków opioidowych
Dihydrokodeina (DHC)
Dihydrokodeina jest pochodną kodeiny, która powstaje poprzez uwodornienie podwójnego wiązania w głównym łańcuchu cząsteczki kodeiny. Metabolizm leku jest podobny do metabolizmu kodeiny, ale w odróżnieniu od niej dihyrokodeina jest lekiem aktywnym. Działa z równą siłą także u pacjentów z uwarunkowanym genetycznie wolnym metabolizmem leków. Maksymalne stężenie w surowicy osiąga po około 3 godzinach od podania, a jej średni okres półtrwania wynosi 3,3-4,5 godziny. Dihydrokodeina jest dostępna w postaci tabletek o kontrolowanym uwalnianiu do stosowania co 12 godzin w dawkach 60 i 90 mg. Maksymalna dawka dobowa wynosi 240 mg.