Hipercholesterolemia rodzinna, wielogenowa, wtórna – kiedy wymagają niestandardowego podejścia?
dr n. med. Marcin Wełnicki1,2
dr n. med. Daniel Śliż1,3
prof. dr hab. n. med. Artur Mamcarz1

dr n. med. Marcin Wełnicki

dr n. med. Daniel Śliż

prof. dr hab. n. med. Artur Mamcarz
Dyslipidemia wielogenowa, rodzinna heterozygotyczna hipercholesterolemia czy wtórne zaburzenia lipidowe występują częściej, niż się powszechnie uważa, a ich diagnostyka i leczenie nie ograniczają się tylko do oznaczenia lipidogramu oraz przepisania dużej dawki silnej statyny.
Wprowadzenie
W sierpniu 2016 r. opublikowano nowe wytyczne w zakresie diagnostyki i terapii zaburzeń lipidowych. W większości dotyczących ich komentarzy przewijają się docelowe wartości stężenia lipoprotein o niskiej gęstości (LDL – low density lipoprotein) w poszczególnych grupach ryzyka, przyjmuje się bowiem za podstawę fakt, że dyslipidemię leczy się statynami. To jednak duże uproszczenie, ponieważ w rzeczywistości zaburzenia lipidowe często powodują dylematy diagnostyczne i terapeutyczne.
Tytuł niniejszego artykułu w pierwszej chwili może wydawać się przewrotny. Cóż bowiem oznacza niestandardowe podejście do leczenia zaburzeń lipidowych? Analiza międzynarodowych wytycznych oraz szeregu publikacji (poglądowych i oryginalnych) dotyczących leczenia dyslipidemii prowadzi do wniosku, że podstawową, stosowaną od lat zasadą jest redukcja stężenia LDL do najniższych wartości za pomocą maksymalnej tolerowanej przez pacjenta dawki statyny. „Lipidowi puryści” w tym miejscu zapewne wysuną zarzut, że liczy się redukcja stężenia LDL per se, a narzędzie jest w tym wypadku kwestią drugorzędną.
W praktyce większość prowadzonych lub konsultowanych w codziennej praktyce pacjentów należy do grupy wysokiego lub bardzo wysokiego ryzyka, wymaga więc stosowania statyny niezależnie od wyjściowego stężenia LDL. Takie postępowanie uznaje się więc za podstawowe, właściwe w większości sytuacji. Istnieją jednak przypadki, w których postępowanie oparte na prostej zasadzie „the lower, the better” okazuje się niewystarczające lub nie jest działaniem, które należy podjąć w pierwszej kolejności. Może się wreszcie okazać, że przyjmowanie statyny będzie niemożliwe lub nieskuteczne. W niniejszym artykule opisano kilka przypadków, w których standardowe postępowanie nie wystarczy. Rodzinna hipercholesterolemia, dyslipidemia wielogenowa oraz wtórne zaburzenia lipidowe to właśnie przykładowe sytuacje, w których można mówić o niekonwencjonalnym podejściu.
Dyslipidemia wielogenowa
Dyslipidemia wielogenowa nie jest właściwą nazwą zaburzenia, które autorzy pracy mają na myśli, jednak dość dobrze podkreśla jego charakter. W aktualnych wytycznych European Society of Cardiology (ESC) tę jednostkę chorobową określa się jako rodzinną złożoną hipercholesterolemię (FCH – familial combined hyperlipidaemia).1
Nie ulega wątpliwości, że jest to zaburzenie uwarunkowane genetycznie, a co za tym idzie – dziedziczne. Istnieje jednak wiele genów, których dysfunkcja prowadzi do zaburzeń gospodarki lipidowej, ponadto fenotyp danego zaburzenia w dużej mierze zależy od czynników środowiskowych. Można by więc powiedzieć, że skłonność do dyslipidemii jest dziedziczna. Problem dotyczy nawet 1% populacji. Zwraca się uwagę na częste współwystępowanie fenotypu typowego dla zespołu metabolicznego (ZM) oraz cukrzycy i FCH. Jednocześnie stężenie poszczególnych frakcji lipidów może być podwyższone w różnym stopniu nawet u członków jednej rodziny i m.in. dlatego rozpoznanie FCH jest ustalane bardzo rzadko.1
Wytyczne wskazują, że jeśli stwierdzi się wczesne występowanie incydentów sercowo-naczyniowych w rodzinie pacjenta oraz podwyższone stężenie triglicerydów (TG) i apolipoproteiny B (ApoB) – odpowiednio >133 mg/dl i >125 mg/dl – sugeruje to FCH.1 Warto zwrócić uwagę, że w wytycznych nie wymieniono stężenia LDL, a oznaczenie ApoB w większości typowych ośrodków jest niemożliwe. Fakt ten należy sam w sobie interpretować jako zupełnie inne podejście diagnostyczne. Stratyfikacja całkowitego ryzyka sercowo-naczyniowego będzie zapewne przeprowadzana na ogólnych zasadach, jednak trzeba pamiętać, że w przypadku pacjentów z grupy niskiego ryzyka sercowo-naczyniowego „toleruje się” stężenie LDL do wartości 190 mg/dl.1 FCH stanowi w tym wypadku dodatkowy czynnik ryzyka, jednak trudny do potwierdzenia, a być może również manifestujący się głównie hipertriglicerydemią. O wskazaniach do farmakoterapii hipertriglicerydemii mówi się tymczasem dopiero przy stężeniu TG >200 mg/dl, i to tylko w przypadku pacjentów z grupy wysokiego lub bardzo wysokiego ryzyka.
Wróćmy do sugestii dotyczących rozpoznania FCH – otóż autorzy aktualnych wytycznych podkreślają znaczenie obciążającego wywiadu rodzinnego, a więc występowania incydentów sercowo-naczyniowych przed 60 r.ż. u kobiet lub przed 55 r.ż. u mężczyzn. Skala SCORE (Systematic Coronary Risk Evaluation) skonstruowana jest dla populacji od 40 r.ż., potencjalnie więc ryzyko wystąpienia zgonu z przyczyn sercowo-naczyniowych dla pacjenta z FCH w tych dwóch krytycznych dekadach może być niedoszacowane, a ryzyko wystąpienia incydentu sercowo-naczyniowego niezakończonego zgonem – z całą pewnością jeszcze wyższe, mimo „jedynie” umiarkowanych lub granicznych nieprawidłowości w lipidogramie. Być może inne klasyczne czynniki ryzyka, takie jak: nikotynizm, płeć męska i nadciśnienie tętnicze, zadecydują o przesunięciu granicy tolerancji stężenia LDL z 190 mg/dl na 155 mg/dl lub nawet 100 mg/dl i przesądzą o wcześniejszym włączeniu typowej strategii postępowania, tj. farmakoterapii statyną.
W przypadku osoby młodszej o dekadę sytuacja jeszcze bardziej się komplikuje. Trzydziestokilkuletni mężczyzna z umiarkowanie podwyższonym stężeniem LDL i TG oraz obniżonym stężeniem cholesterolu frakcji lipoprotein o dużej gęstości (HDL – high density lipoprotein) ma prawdopodobnie ZM. Prawidłowe oszacowanie całkowitego ryzyka sercowo-naczyniowego takiej osoby może być bardzo trudne. Pacjent nie cierpi jeszcze na cukrzycę, ma natomiast otyłość brzuszną, skutecznie leczone nadciśnienie tętnicze i typowe dla ZM zaburzenia lipidowe. W zdecydowanej większości laboratoriów stężenie LDL nie jest oznaczane bezpośrednio, tylko wyliczane z formuły Friedewalda. Wzór ten zaniża stężenie LDL przy wysokich stężeniach TG, a istotne zafałszowanie wyników może nastąpić już przy stężeniach TG ok. 200 mg/dl. Wydaje się więc, iż szczególnie w przypadku pacjentów z ZM i wywiadem rodzinnym obciążonym przedwczesnym występowaniem incydentów sercowo-naczyniowych diagnostykę warto uzupełnić o oznaczenie stężenia ApoB i cholesterolu nie-HDL oraz pomiar obwodu talii. Według aktualnych wytycznych stężenie ApoB i cholesterolu nie-HDL powinno być rozważane jako drugorzędowy cel terapeutyczny (klasa zalecenia IIa, poziom dowodu B dla obydwu oznaczeń), z kolei obwód talii oraz stężenie TG wydają się prostymi wskaźnikami podwyższonego ryzyka sercowo-naczyniowego u pacjentów z ZM.1 Oczywiście kluczowym elementem postępowania, bazą potencjalnej farmakoterapii, będzie zmiana nawyków żywieniowych i zwiększenie aktywności fizycznej. Zakładając jednak, że u pacjenta rzeczywiście mamy do czynienia z FCH, skuteczność postępowania niefarmakologicznego będzie prawdopodobnie umiarkowana.
Niestandardowość postępowania w tym przypadku polega więc przede wszystkim na wysunięciu podejrzenia FCH, rozszerzeniu diagnostyki biochemicznej o oznaczenia, które nie są powszechnie dostępne, oraz na rozważeniu wcześniejszego włączenia leczenia farmakologicznego, niż sugerowałaby analiza klasycznych czynników ryzyka.
Rodzinna hipercholesterolemia
Heterozygotyczna rodzinna hipercholesterolemia (HeFH – heterozygous familial hypercholesterolaemia) to najczęściej spotykane, monogenowe zaburzenie gospodarki lipidowej.1,2 HeFH w ponad 90% przypadków jest spowodowana mutacją genu odpowiadającego za receptory dla LDL (LDLR, mutacja o typie utraty funkcji), zdecydowanie rzadziej uszkodzony jest gen kodujący ApoB, najrzadziej zaś – gen kodujący proproteinową konwertazę subtylizyny/keksyny 9 (PCSK9 – proprotein convertase subtilisin-like kexin).3,4 Do niedawna uważano, że na rodzinną hipercholesterolemię choruje ok. 1 na 500 osób, obecnie jednak sugeruje się, że występuje ona znacznie częściej: u 1 na 200-250 osób.3
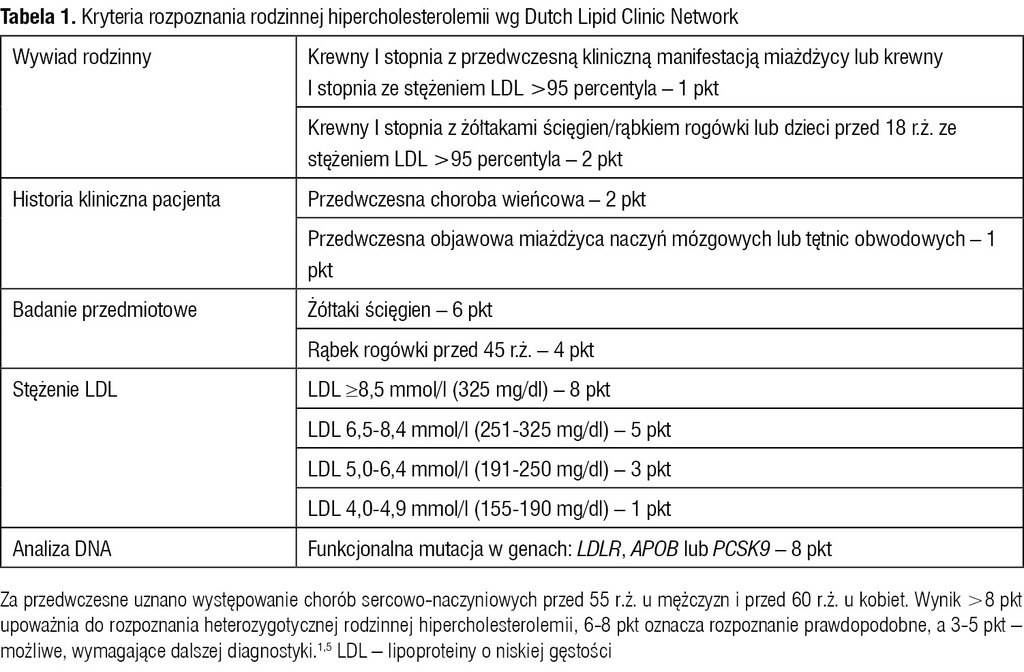
Tabela 1. Kryteria rozpoznania rodzinnej hipercholesterolemii wg Dutch Lipid Clinic Network
Typowym zaburzeniem biochemicznym dla tej choroby jest wysokie lub bardzo wysokie całkowite stężenie cholesterolu i frakcji LDL. Stężenie LDL najczęściej przekracza 200 mg/dl, nierzadko 300 mg/dl, jednak nawet stężenie <190 mg/dl nie wyklucza HeFH.1 Warto podkreślić, że w większości krajów na świecie rozpoznaje się <1% HeFH. W Europie wyróżniają się pod tym względem Holandia (ok. 70% rozpoznanych przypadków), Norwegia (ok. 40%) i Islandia (ok. 19%), w krajach takich jak Szwajcaria czy Anglia rozpoznaje się już <15% przypadków HeHF, w pozostałych krajach zaś <10%.3,5 Ustalenie prawidłowego rozpoznania jest bardzo istotne. Ryzyko wystąpienia choroby wieńcowej u osoby, u której rozpoznanie HeFH zostanie potwierdzone na podstawie kryteriów Dutch Lipid Clinic Network (tab. 1), jest 10- lub 13-krotnie wyższe od populacyjnego zależnie od tego, czy osoba ta przyjmuje statyny.3,5
Podstawowe metody postępowania w przypadku rozpoznania HeFH nie różnią się od zasad leczenia dyslipidemii nieuwarunkowanej genetycznie. Autorzy aktualnych wytycznych podkreślają jednak konieczność agresywnego leczenia wysokimi dawkami statyn pacjentów dorosłych.1