Potranskrypcyjne mechanizmy regulacji COX-2
Ostatnio przeprowadzone badania wykazały, że regulacja genu COX-2 odbywa się głównie na poziomie potranskrypcyjnym.14 Szczególnie ważny mechanizm regulacji COX-2 reprezentowany jest przez niepodlegający translacji region 3’ (3’-UTR) COX-2, który może wpływać na stabilność mRNA i wydajność procesu translacji.15 Region ten zawiera wiele kopii elementów bogatych w adeninę i uracyl (AU-rich) (ARE) zbudowanych z sekwencji 5’-AUUUA-3’. Elementy te, występujące także w obrębie 3’-UTR mRNA wielu protoonkogenów i cytokin, zapewniają potranskrypcyjną kontrolę ekspresji, działając jako determinanty niestabilności mRNA lub jako składowa hamująca translację.16
Wiele grup badawczych wykryło polipeptydy swoiście oddziałujące z ARE.17 Do tych regulujących czynników należy wiele białek wiążących cytoplazmatyczny mRNA, zaangażowanych w destabilizację, stabilizację i przetwarzanie mRNA oraz transport jądrowo-cytoplazmatyczny. Opisano wiele białek wiążących mRNA i rozpoznających ARE, aczkolwiek mało poznane są mechanizmy, za pośrednictwem których czynniki te wpływają na degradację mRNA oraz hamowanie translacyjne.
Obecnie prowadzone są badania nad rolą tych białek w potranskrypcyjnej kontroli COX-2. Sugeruje się w szczególności, że HuR – białko wiążące się z mRNA należące do rodziny ELAV (embryonic lethal abnormal vision) – może prowadzić do zwiększenia ekspresji COX-2. Wiązanie innych białek, takich jak TIA-1,18 TIAR, oraz tristetraproliny19 zmniejsza ekspresję COX-2. Inne białka, jak AUF-1, także mogą odgrywać rolę zarówno w degradacji, jak i stabilizacji mRNA kodującego COX-2. W końcu wykazano,20 że CUGBP2 hamuje translację mRNA dla COX-2 mimo spowolnienia jego degradacji.
Począwszy od pojawienia się danych z badań in vitro, które dowodzą, że HuR zmniejsza nadmierną ekspresję białka COX-2, przez przyłączanie do ARE mRNA dla COX-2 zaprojektowaliśmy badanie21 mające na celu sprawdzenie hipotezy o modulującym wpływie HuR na ekspresję mRNA dla COX-2 w przypadku niestabilności blaszki miażdżycowej. Analizowaliśmy ekspresję HuR w blaszkach miażdżycowych tętnic szyjnych u bezobjawowych i objawowych pacjentów, odnosząc to do ekspresji COX-2 i klinicznych wykładników destabilizacji blaszki miażdżycowej. HuR i COX-2 charakteryzowały się większą ekspresją w blaszkach miażdżycowych tętnic szyjnych pacjentów objawowych. Co ciekawe, zarówno badania immunohistochemiczne w skrawkach blaszek miażdżycowych, jak i immunofluorescencyjne z podwójnym znakowaniem wykazały silny związek między ekspresją Hur i COX-2. Dlatego badanie to wydaje się potwierdzać, że HuR może przyczyniać się do niestabilności blaszki miażdżycowej w tętnicach szyjnych w wyniku opóźnienia degradacji mRNA dla COX-2.
Zło wcielone: COX-2 czynnościowo związane z syntazą mPGE
Obecnie uważa się, że w patogenezie aterotrombozy zmniejszenie zawartości elementów macierzy czapeczki włóknistej – szczególnie kolagenu fibrylarnego – spowodowane nierównowagą między procesami syntezy i rozpadu, prowadzi do jej scieńczenia. To z kolei naraża czapeczkę włóknistą na samoistne pęknięcie pod wpływem czynników hemodynamicznych lub innych bodźców.22 Odpowiedzialność za nasilone procesy rozpadu przypisuje się rodzinie metaloproteinaz degradujących macierz, w szczególności MMP-1, MMP-2, MMP-3 i MMP-9, które występują w komórkach zapalnych blaszek miażdżycowych (makrofagi, komórki piankowate) oraz w mniejszym stopniu w komórkach mięśni gładkich i komórkach śródbłonka.23
Warto nadmienić, że synteza MMP-2 i MMP-9 przez makrofagi odbywa się szlakiem zależnym od PGE2/cyklicznego adenozynomonofosforanu (cAMP).24 Przekazywanie sygnału w tym szlaku modulowane jest przez COX-2 i syntazę PGE (PGES). Opisano trzy izoformy PGES: cytozolową PGES (cPGES), oraz typ 1 i typ 2 mikrosomalnej PGES (mPGES). O ile cPGES i w dużym stopniu mPGES-2 występują konstytutywnie, o tyle mPGES-1 pojawia się w odpowiedzi na bodźce zapalne, co wskazuje, że enzym ten zaangażowany jest w tworzenie prostaglandyn w przebiegu chorób zapalnych.25
Prawdopodobieństwo, że wywołana zapaleniem jednoczesna indukcja COX-2 oraz mPGES-1 może być mechanizmem uszkadzającym płytkę miażdżycową, skłoniło nas do zbadania,26 czy wpływa to na produkcję metaloproteinaz macierzy przez makrofagi w ludzkich płytkach miażdżycowych. Dane wskazują na zwiększoną ekspresję aktywnej enzymatycznie MMP-2 i MMP-9 przez makrofagi w podatnych na uszkodzenie regionach niestabilnych blaszek miażdżycowych, najprawdopodobniej z powodu nasilenia syntezy PGE2 wywołanej indukcją czynnościowo połączonych COX-2/mPGES-1. Dlatego też miejscowy wzrost aktywności metaloproteinaz macierzy zależnych od PGE2 potencjalnie przyczynia się do pęknięcia blaszki miażdżycowej.
Patofizjologiczną rolę czynnościowo połączonych COX-2/ /mPGES-1 potwierdzają także badania wskazujące, że ekspresja mPGES-1 jest istotnie zwiększona przez czynniki prozapalne w komórkach ściany naczyniowej, a ulega zmniejszeniu pod wpływem deksametazonu27 przy odpowiednich zmianach ekspresji COX-2 i opóźnionej generacji PGE2. Dlatego w procesie miażdżycowym u ludzi nadmierna ekspresja funkcjonalnie połączonych COX-2/mPGES-1 w makrofagach może stanowić dominujący szlak przemian kwasu arachidonowego, prowadzący do zwiększonej biosyntezy PGE2 i MMP zależnych od PGE2.
Z tego względu zwiększenie aktywności COX-2, czynnościowo połączonej z prozapalną mPGES-1, jest kluczowym enzymem w procesie rozwoju niestabilnej blaszki miażdżycowej.
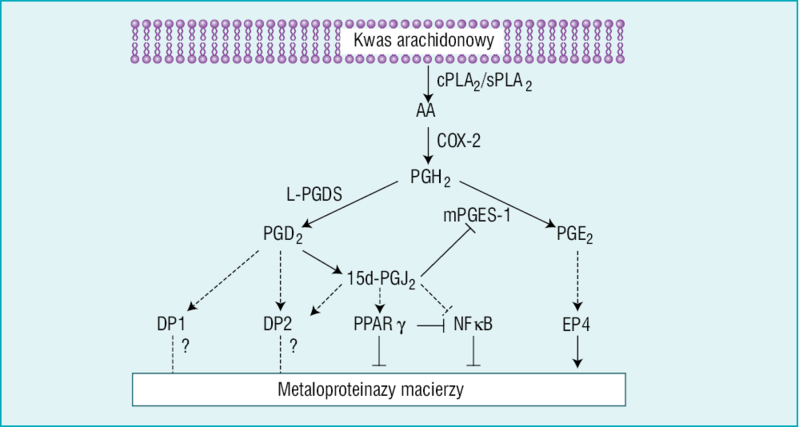
Rycina 1. Szlak przemian kwasu arachidonowego (AA) – reakcje katalizowane przez syntazę prostaglandyny D typu lipokaliny (L-PGDS) i typ 1 mikrosomalnej syntazy prostaglandyny E (mPGES-1) oraz wpływ metabolitów wyżej wymienionych szlaków na ekspresję metaloproteinaz macierzy (MMP). Pod wpływem L-PGDS i mPGES-1 prostaglandyna (PG) H2 może zostać przekształcona odpowiednio w PGD2 i PGE2. PGD2 przekształca się do 15-deoxy, δ12-14 (15d)-PGJ2, która z kolei hamuje ekspresję MMP poprzez blokowanie receptora aktywowanego proliferatorami peroksysomów γ (PPARγ), czynnika jądrowego (NF) κB oraz mPGES-1. Ciągle niejasne jest, czy 15d-PGJ2, poprzez receptor DP2 dla PGD2, może wpływać na tworzenie MMP. Nie wiadomo także, czy PGD2 może wpływać na biosyntezę MMP za pośrednictwem swoich receptorów DP1 i DP2. PGE2 wytworzona przez mPGES-1 promuje tworzenie MMP poprzez związany z białkiem G heptahelikalny receptor EP4. COX-2 – cyklooksygenaza 2; cPLA2 – cytozolowa fosfolipaza A2; sPLA2 – wydzielnicza fosfolipaza A2
Dodatkowym aspektem złożoności tego zagadnienia jest fakt, że istnieje więcej niż jeden receptor, który pośredniczy w działaniu PGE2. PGE2 może działać za pośrednictwem co najmniej czterech różnych receptorów heptahelikalnych (EP1-4) związanych z białkami G. Ostatnio zasugerowano, że EP4 może być głównym receptorem zaangażowanym w patofizjologię uwalniania MMP (ryc. 1).28 McCoy i wsp.28 w badaniach na myszach pozbawionych każdego spośród czterech znanych receptorów EP wykazali, że tylko myszy EP4–/– charakteryzują się znaczną opornością na rozwój eksperymentalnego reumatoidalnego zapalenia stawów, a w próbkach z ich kości stwierdzono istotnie mniejszą ekspresję MMP-2 niż u myszy EP4+/+ leczonych za pomocą PGE2.29
Stąd prawdopodobieństwo, że nadmierna ekspresja jednego swoistego receptora dla PGE2 może wpływać na mechanizm zależnej od PGE2 destabilizacji blaszki miażdżycowej, co z kolei skłoniło nas do zbadania,30 czy jeden z czterech receptorów EP może swoiście modulować produkcję MMP przez makrofagi blaszek miażdżycowych. Dowiedliśmy,30 że receptor EP-4 zaangażowany jest w zależną od PGE2 nadmierną ekspresję MMP w blaszkach miażdżycowych wywołujących objawy kliniczne. Zanotowaliśmy większą ekspresję EP4 w próbkach z tętnic szyjnych pacjentów z niedawno przebytym udarem mózgu niż w próbkach pochodzących od pacjentów bezobjawowych.
Co więcej, ponieważ wykazano,31 że PGE2 może wzmacniać ekspresję COX-2 w komórkach zapalnych w mechanizmie zależnym od EP-4, nadmierna ekspresja EP-4 w makrofagach blaszek miażdżycowych tętnic szyjnych może wywołać samonapędzający się autokrynny i parakrynny mechanizm sprzężenia zwrotnego, wzmacniający i przedłużający reakcję zapalną zależną od COX-2, prowadząc do postępującej destabilizacji blaszki miażdżycowej (ryc. 1). Obecnie, kiedy EP4 wydaje się być unikalnym receptorem zaangażowanym w destabilizujące działanie PGE-2, jego blokada może być idealnym celem działań zapewniających stabilizację blaszki miażdżycowej.
Anioł: COX-2 czynnościowo połączona z PGI2 oraz syntazą PGD2
Zaniepokojenie niekorzystnym wpływem koksybów na układ krążenia zogniskowało uwagę środowiska naukowego na protekcyjnej roli COX-2 występującej w komórkach śródbłonka i czynnościowo połączonej z syntazą PGI2. Jednoznacznie wykazano, że PGI2 nie jest wyłącznie silnym związkiem rozurczającym naczynia i antyagregacyjnym, ale także ma działanie przeciwzapalne i przeciwmiażdżycowe.32 W odróżnieniu od PGI2, w procesie aterotrombozy mniej jasna (lub bardziej złożona) jest rola PGD2.
COX-2 jest tylko pośrednim enzymem w szlaku metabolicznym kwasu arachidonowego, a produkt COX-2, jakim jest PGH2, jest dalej metabolizowany przez inne izomerazy do różnych prostanoidów (PGE2, PGD2, PGF2α, PGI2, tromboksan A2). Dlatego też stosunkowo duża ilość jednego swoistego prostanoidu w porównaniu z innymi jest skutkiem ekspresji i aktywności swoistej izomerazy, i jedynie współistniejąca ekspresja czynnościowo połączonych COX-2 i PGES może prowadzić do biosyntezy metaloproteinaz macierzy zależnych od PGE2 w obrębie blaszki miażdżycowej (ryc. 1). Jest to szczególnie ważne, biorąc pod uwagę, że prostaglandyny, podobnie jak krążąca syntaza PGD (PGDS), mają właściwości przeciwzapalne (ryc. 1).
Krążąca PGDS okazała się być czynnikiem chroniącym przed restenozą u pacjentów poddawanych przezskórnej angioplastyce naczyń wieńcowych. Co więcej, wykazano, że pochodna PGDS – 15-deoxy, δ12-14-PGJ2 (15d-PGJ2) – jest silnym inhibitorem czynnika jądrowego κB (NF-κB)33 oraz IκK,34 i że poprzez mechanizm obejmujący pobudzenie receptorów aktywowanych proliferatorami peroksysomów γ (PPARγ) może już w ilościach pikogramowych działać przeciwzapalnie (ryc. 1).35 W świetle tego należy pamiętać, że przy wykorzystaniu metod barwienia wykryto 15d-PGJ2 w makrofagach ludzkich tętnic szyjnych wraz z ekspresją COX-2.36
Istnieją dwie różne izoformy PGDS. Pierwszą jest krwiopochodna PGDS (H-PGDS), drugą zaś PGDS typu lipokaliny (L-PGDS), która występuje w blaszkach miażdżycowych u ludzi.37 Warto wspomnieć, że czynniki prozapalne, takie jak TNFα wykryty w zmianach miażdżycowych, zmniejszają syntezę PGD2 oraz zwiększają uwalnianie PGE2,38 podczas gdy glukokortykoidy mają działanie przeciwne.39 Dlatego też stosunkowo duża ilość swoistych prostanoidów jest skutkiem ekspresji i aktywności swoistych izomeraz, zaś dominująca ekspresja mPGES-1 w porównaniu z L-PGDS może prowadzić do nasilenia procesu zapalnego oraz uszkodzenia blaszki miażdżycowej w obrębie tętnic szyjnych.