Układowe zapalenia małych naczyń
Znacznie częściej niż w chorobie Behçeta do uszkodzenia obwodowego układu nerwowego dochodzi w przebiegu układowych zapaleń małych naczyń. Neuropatia obwodowa jest najczęstszym (i długo może być jedynym) objawem choroby. Martwicze zapalenie naczyń powoduje niedokrwienie nerwów, co prowadzi do ich uszkodzenia. Neuropatia może mieć charakter ostry lub wolno postępujący. Najczęstszą manifestacją jest wieloogniskowa mononeuropatia (ponad 60%), która dotyczy nerwu strzałkowego (89%), nerwu łydkowego (84%), nerwu piszczelowego, łokciowego oraz nerwu pośrodkowego. Druga co do częstości jest symetryczna polineuropatia obwodowa, stwierdzana u mniej niż ⅓ pacjentów. Zwykle dotyczy ona dystalnego odcinka kończyn dolnych. Rzadko występuje asymetryczna neuropatia czuciowa. Często obserwuje się też zajęcie nerwów czaszkowych, na ogół twarzowego, rzadziej III lub VI. Spośród wszystkich neuropatii obwodowych neuropatie w przebiegu chorób układowych (SVN – systemic vasculitic neuropathy) występują u 85% chorych, w tym 60% w przebiegu pierwotnych układowych zapaleń naczyń, a pozostałe 15% stanowią neuropatie idiopatyczne (NSVN – non-systemic vasculitic neuropathy).8,9
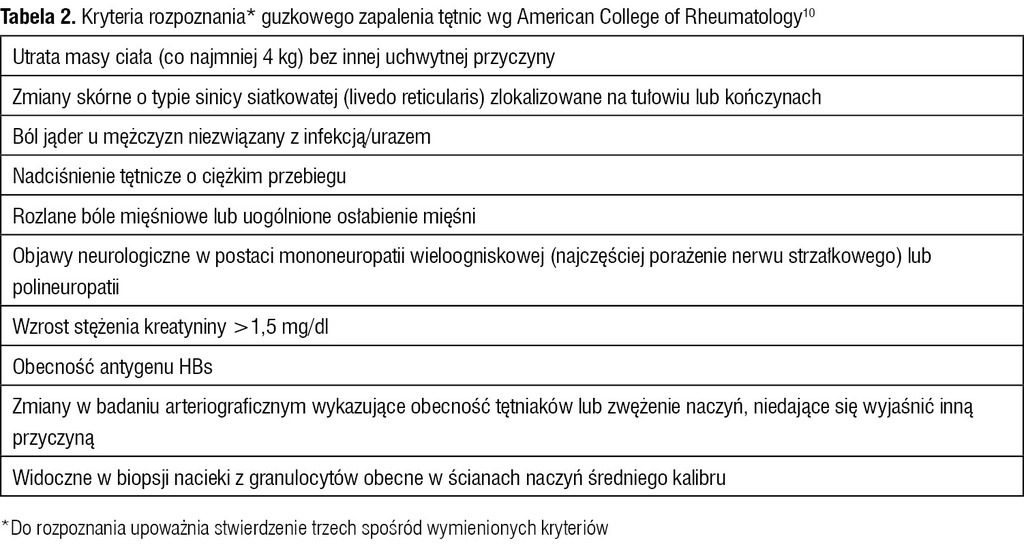
Tabela 2. Kryteria rozpoznania* guzkowego zapalenia tętnic wg American College of Rheumatology10
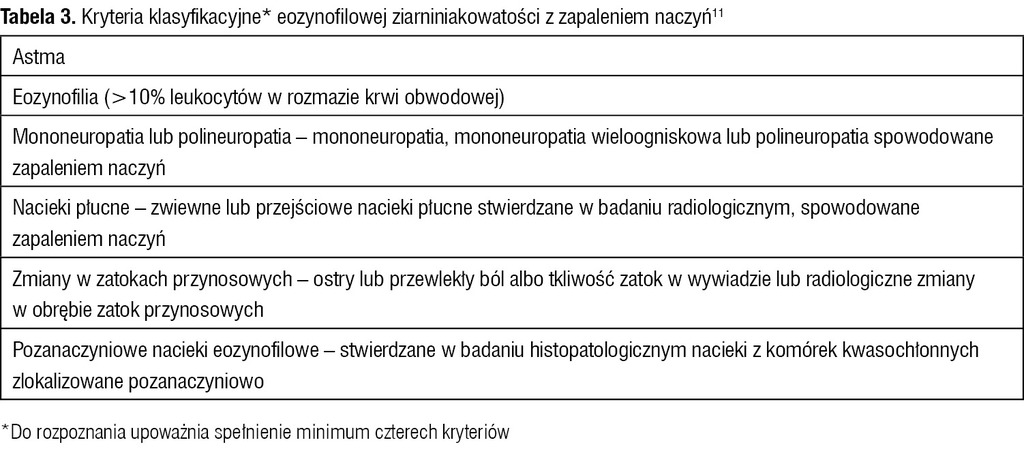
Tabela 3. Kryteria klasyfikacyjne* eozynofilowej ziarniniakowatości z zapaleniem naczyń11
Według American College of Rheumatology (ACR) objawy zajęcia obwodowego układu nerwowego są jednym z kryteriów rozpoznania guzkowego zapalenia tętnic (PAN – polyarteritis nodosa) (tab. 2) oraz eozynofilowej ziarniniakowatości z zapaleniem naczyń (EGPA – eosinophilic granulomatosis with polyangiitis), czyli zespołu Churga-Strauss (tab. 3). Objawy te występują u 60-85% chorych z EGPA oraz PAN. Z reguły przyjmują postać mononeuropatii mnogiej, poza tym dystalnej symetrycznej lub niesymetrycznej polineuropatii oraz neuropatii skórnej.
Zwykle pierwszym objawem neuropatii jest rozlany ból (ból stanowi objaw towarzyszący u ponad 80% chorych), np. w kończynie. W dalszej kolejności następuje osłabienie uszkodzonego nerwu. Około 75% pacjentów doświadcza co najmniej jednego ostrego ataku, tylko u ok. 25% choroba ma powolny, stopniowo postępujący przebieg. Typowe jest nagłe pojawienie się niedowładów lub porażeń. Niespecyficzne objawy ogólne, takie jak: utrata wagi, ogólne osłabienie, gorączki, stwierdza się u ok. 80% chorych z neuropatią o etiologii związanej z układowym zapaleniem naczyń. Do zajęcia OUN w EGPA oraz w PAN dochodzi jedynie w ok. 5% przypadków, zwykle w trakcie trwania choroby, od 2 do 3 lat od początku objawów. Do najczęstszych manifestacji zaliczają się przemijające niedokrwienie mózgu (TIA – transient ischemic attack) i zaniewidzenie jednooczne.

Tabela 4. Kryteria klasyfikacyjne* ziarniniakowatości z zapaleniem naczyń12
Drugi typ objawów jest związany z rozlaną encefalopatią, której towarzyszą napady padaczkowe i nagłe pogorszenie stanu świadomości. Zmiany ogniskowe mogą pojawić się nagle jako następstwo obszarów niedokrwiennych w obrębie kory, pnia mózgu i móżdżku. Według różnych autorów objawy neurologiczne w przebiegu ziarniniakowatości z zapaleniem naczyń (GPA – granulomatosis with polyangiitis), dawniej ziarniniakowatości Wegenera (kryteria rozpoznania zebrano w tabeli 4), występują w 25% do nawet 50% przypadków. Przed erą leczenia cyklofosfamidem były znacznie częstsze niż obecnie, rzadko stanowią pierwszy objaw choroby. Zwykle w przebiegu choroby obserwuje się obwodowe neuropatie (najczęściej ostrą mononeuropatię mnogą) oraz uszkodzenia nerwów czaszkowych, rzadziej: drgawki, udar czy zapalenie mózgu. Neuropatie nerwów czaszkowych (zwykle II, VI, VII) są spowodowane po pierwsze zapaleniem naczyń odżywczych nerwów, po drugie – bezpośrednim uciskiem ziarniny zapalnej i obrzękiem otaczających tkanek.13
Opisane wyżej dwa typy zapaleń małych naczyń krwionośnych (EGPA i GPA) zaliczane są do grupy zapaleń naczyń zależnych od przeciwciał przeciw cytoplazmie granulocytów obojętnochłonnych (neutrofilów) (ANCA – anti-neutrophil cytoplasmic antibodies). PAN rozwija się na skutek uszkodzenia naczyń przez tworzące się kompleksy immunologiczne.
Toczeń rumieniowaty układowy
SLE, RZS oraz zespół Sjögrena są trzema najczęściej występującymi chorobami reumatycznymi. U chorych nimi dotkniętych stwierdza się różne neurologiczne komplikacje, które powodują wzrost śmiertelności oraz pogarszają rokowanie, a często bywają także pierwszym objawem choroby podstawowej.
Patogeneza zmian neurologicznych w SLE pozostaje niejasna. Do najczęściej opisywanych nieprawidłowości w materiale histopatologicznym mózgu chorych na SLE należą mikroudary. Zwykle są one asymptomatyczne klinicznie, nie korelują z objawami endocarditis czy zakrzepicą. Nie znaleziono też dowodów na zapalenie naczyń mózgowych. Możliwe, że uszkodzenie systemu nerwowego wynika z bezpośredniego działania przeciwciał i cytokin zapalnych. Na przykład przeciwciała antyfosfolipidowe (APL – antiphospholipid antibodies) wpływają zarówno na waskulopatie małych naczyń, jak i zakrzepicę w dużych naczyniach. Uważa się, że przeciwciała antyneuronalne oznaczane w płynie mózgowo-rdzeniowym, szczególnie przeciwciała przeciwko podjednostce NR2 receptora N-metylo-D-asparaginianu (NMDA – N-methyl-D-aspartate), mają związek z objawami neuropsychiatrycznymi, a przeciwciała przeciwko podjednostce NR1 receptora NMDA – z zapaleniem mózgu u tych chorych. Przeciwciała antyrybosomalne początkowo były uważane za marker neuropsychiatrycznego zajęcia obwodowego układu nerwowego, lecz ich czułość wynosi 26%, a specyficzność 80%, co znacząco ogranicza ich użyteczność kliniczną. Wzrost stężenia cytokin: IL6 i interferonu łączy się ze wzrostem ryzyka psychoz i drgawek.
Postać neuropsychiatryczna tocznia występuje w 14-75% przypadków. U ok. 43% chorych objawy pojawiają się przed ustaleniem rozpoznania SLE, a u 63% – w pierwszym roku od rozpoznania. Do najczęstszych zaburzeń neuropsychiatrycznych w SLE należą: bóle głowy (28-57%), zaburzenia nastroju (21%), dysfunkcje poznawcze (20%), drgawki (10%) i choroby naczyń mózgowych (8%). Chociaż bóle głowy stanowią najczęstszy objaw neurologiczny w toczniu, nie są częstsze niż w populacji ogólnej. Podobnie depresja w toczniu występuje równie często jak w populacji ludzi zdrowych.
Psychozy są rzadsze (ok. 11%) i związane z aktywnością choroby. Zwykle obejmują omamy i myśli paranoiczne. Psychozy mogą być pierwszym objawem choroby. Znacznie częściej niż psychozy obserwuje się u tych chorych zaburzenia funkcji poznawczych. W standardowych testach neuropsychiatrycznych, które stosowano w wielu dużych badaniach klinicznych, stwierdzono je u 60-80% pacjentów. Zwykle zaburzone są umiejętności wzrokowo-przestrzenne (np. rozpoznawanie kształtów, ocena odległości) oraz pamięć wzrokowa i werbalna. Obecność przeciwciał przeciwko receptorowi NMDA i przeciwciał antykardiolipinowych (ACL – anticardiolipin antibodies) w klasie immunoglobuliny M (IgM) oraz wysokie miano przeciwciał przeciwjądrowych (ANA – antinuclear antibodies) wiążą się z większym ryzykiem występowania zaburzeń poznawczych.
Drgawki mogą pojawiać się zarówno przed rozwojem innych objawów choroby, jak i podczas jej trwania. Czynniki predysponujące do drgawek to przebyty udar niedokrwienny, obecność antykoagulantu toczniowego (LA – lupus anticoagulant), APL i przeciwciał anty-Sm. Zwykle mamy do czynienia z pojedynczymi epizodami napadów drgawkowych, ale wysoka aktywność choroby predysponuje do nawracających i przedłużających się stanów drgawkowych. W SLE częściej niż w populacji ogólnej obserwowano udary niedokrwienne. Oprócz konwencjonalnych czynników ryzyka udaru (nadciśnienie, dyslipidemia, cukrzyca) – częstych u chorych na SLE, wynikających w dużej mierze z przedłużającego się leczenia glikokortykosteroidami oraz prowadzących do przyspieszonej miażdżycy – predykatorem występowania udaru są także APL. U 65% chorych na SLE z udarem stwierdzono występowanie tych przeciwciał.14-16

Tabela 5. Objawy neuropsychiatryczne możliwe w toczniu rumieniowatym układowym
Wszystkie objawy neuropsychiatryczne możliwe w SLE zebrano w tabeli 5.
Zespół antyfosfolipidowy
Badania wykazały, że u 50% chorych na SLE, u których stwierdzono obecność ACL, w ciągu 10 lat rozwija się wtórny APS. Wiąże się to z ryzykiem powikłań zakrzepowych i wpływa na wzrost śmiertelności w tej grupie chorych. APS może być pierwotny lub wtórny do innych chorób, w jego przebiegu występują zakrzepica w naczyniach żylnych i/lub tętniczych oraz powikłania w trakcie ciąży, którym zawsze towarzyszy obecność APL. APL stwierdzane są u ok. 1-3% dorosłej populacji bez klinicznych objawów APS, u 25% chorych z zakrzepicą żylną, u 25% młodych chorych z udarem, u 18% chorych z przedwczesną miażdżycą naczyń wieńcowych oraz u ok. 30% pacjentek z nawracającymi utratami ciąż. Udary niedokrwienne w przebiegu APS dotykają częściej młodszych pacjentów w porównaniu z tymi bez APS. Często zgłaszanym objawem są migrenowe bóle głowy, ponadto opisywano upośledzenie funkcji poznawczych (od łagodnych do ciężkich zespołów otępiennych pochodzenia naczyniowego) oraz napady drgawkowe, które ściśle wiążą się z obecnością APL. U niektórych pacjentów z APS występują objawy przypominające stwardnienie rozsiane (pseudo-SM; SM – sclerosis multiplex). U tych chorych częściej zdarzają się ogniskowe epizody niedokrwienne w mózgu: przeważnie TIA, rzadziej (zwykle u dzieci i młodych kobiet) obserwowano pląsawicę, mielopatię poprzeczną czy objawy ze strony narządu wzroku.
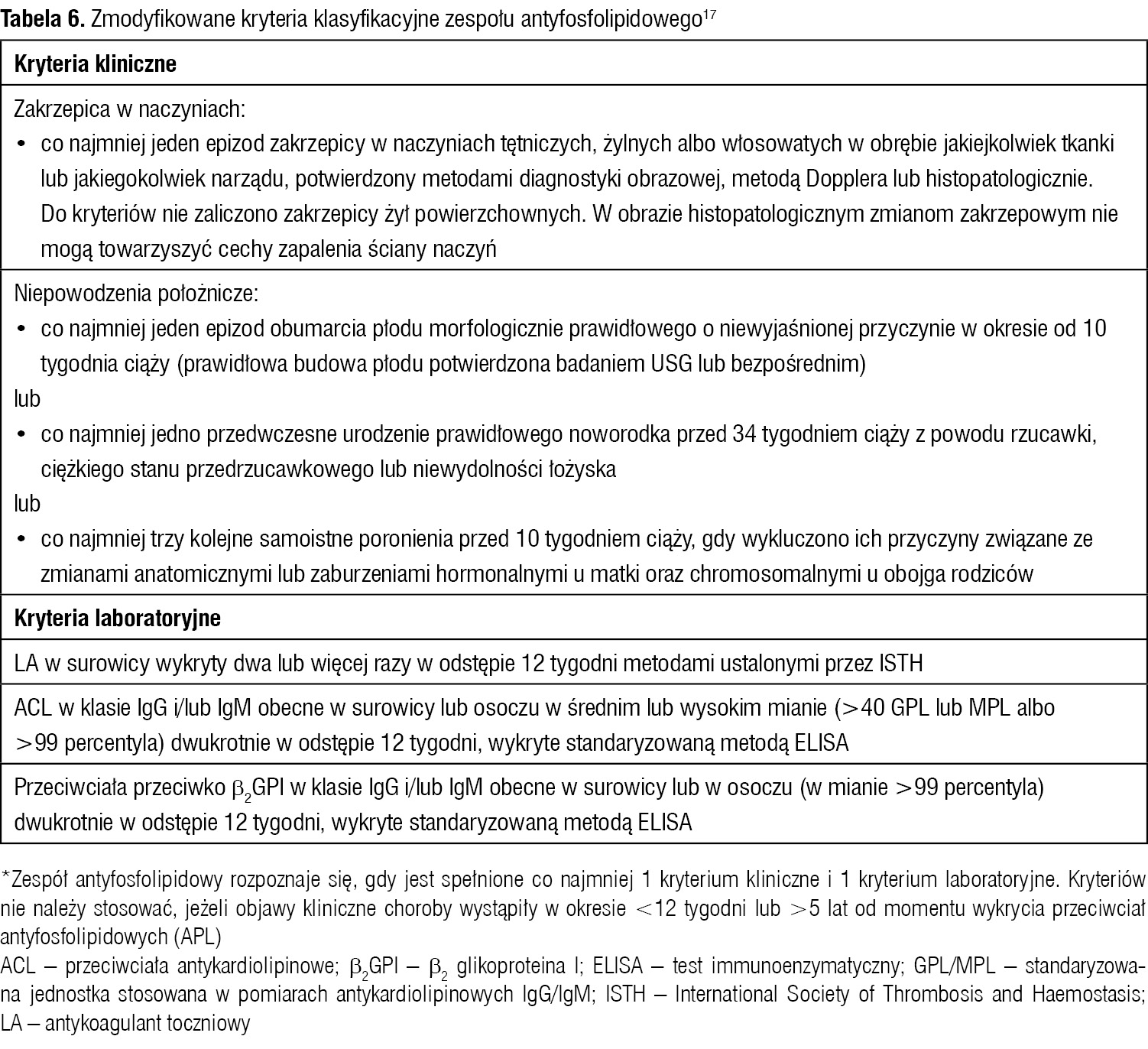
Tabela 6. Zmodyfikowane kryteria klasyfikacyjne zespołu antyfosfolipidowego17