Astrocyty są najliczniejszymi komórkami glejowymi mózgu i pełnią wiele ważnych funkcji, w tym strukturalne, odżywcze, naprawcze, a także regulujące przepływ krwi (przez tzw. falę Ca++) i modulujące neuroprzekaźnictwo. Biorą udział w tworzeniu bariery krew–mózg. Otaczają synapsy, uczestnicząc w przemianie neurotransmiterów, zwłaszcza glutaminianów i kwasu γ-aminomasłowego (GABA – γ-aminobutyric acid). Pośredniczą także w wymianie różnych składników między naczyniami krwionośnymi a tkanką mózgową, wydzielają kilka czynników wzrostowych oraz mają znaczenie w utrzymaniu równowagi wodno-elektrolitowej i stałości środowiska tkanki mózgowej. Ich wypustki otaczają przewężenia Ranviera, zapewniając izolację konieczną do szybkości przewodzenia bodźca elektrycznego. Ostatnio pojawiły się doniesienia, że odgrywają też rolę w realizacji funkcji poznawczych. W warunkach fizjologicznych w istocie białej przeważają astrocyty włókniste.5
Komórki mikrogleju to specyficzne dla OUN komórki pochodzenia mezenchymalnego, biorące udział w odpowiedzi immunologicznej, kontrolujące homeostazę oraz będące źródłem makrofagów mózgowych i cytokin.
Patogeneza leukodystrofii i genetycznie uwarunkowanych leukoencefalopatii
Patogeneza LD i gLE jest bardzo złożona i różna dla każdej z jednostek chorobowych i podgrup chorób. Jak wspomniano wyżej, u podłoża LD leżą zarówno niedobory białek biorących udział w budowie, odnawianiu oraz funkcjonowaniu mieliny, jak i defekty cząsteczek RNA uczestniczących w metabolizmie podstawowym komórek. Patologia może również wynikać z uszkodzenia przez różne spichrzane metabolity lub endogenne substancje toksyczne (np. psychozyna w chorobie Krabbego). W wielu współistnieje czynnik zapalny, np. udział cytokin pochodzących z mikrogleju w patogenezie adrenoleukodystrofii sprzężonej z chromosomem X (X-ALD – X-linked adrenoleukodystrophy), a także w chorobie Krabbego.1
W gLE uszkodzenie może wynikać z: 1. patologii naczyń (np. mózgowa dominująca arteriopatia z udarami podkorowymi, leukoencefalopatią i otępieniem [CADASIL – cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy], arteriopatia mózgowa dziedziczona autosomalnie recesywnie z zawałami podkorowymi i encefalopatią [CARASIL – cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy], choroba Fabry’ego); 2. toksycznego działania produktów przemian pośrednich (np. aminoacydopatie, kwasice organiczne); 3. zmian w istocie pozakomórkowej (np. choroba ze spichrzaniem kwasu sjalowego i niektóre rzadkie kolagenopatie); 4. zaburzonej odnowy DNA (np. zespół 4H), 5. zaburzeń energetycznych (choroby mitochondrialne) i innych.1,3,4
Diagnostyka leukodystrofii
W obrazie klinicznym LD i gLE zwraca uwagę przewaga zaburzeń ruchowych w początkowym okresie choroby. Z reguły jest tak, że im wcześniej wystąpi choroba, tym cięższy i bardziej nieswoisty ma przebieg.
W chorobach z hipomielinizacją (HLD) przebieg kliniczny jest powolniejszy. Rozwój psychoruchowy, zwłaszcza ruchowy, zwykle jest opóźniony, a u chorych najczęściej stwierdza się uogólnioną wiotkość przy zachowanych odruchach. Prawie zawsze występuje oczopląs. Niekiedy obserwuje się okresy pozornej poprawy i długie fazy plateau, co może się przyczynić do błędnego rozpoznania mózgowego porażenia dziecięcego. Rozwój emocjonalny, społeczny i intelektualny jest względnie lepszy, a niekiedy prawidłowy.11,12 Z kolei w leukodystrofiach demielinizacyjnych (LDD) objawy występują z reguły po okresie prawidłowego rozwoju i mają szybszy przebieg. Zależnie od wieku obserwuje się w nich regres rozwoju/otępienie, ataksję i spastyczność. Współistnienie neuropatii obwodowej może powodować przejściową wiotkość.
W różnicowaniu bywa pomocna analiza okoliczności wystąpienia choroby: początek prawie nieuchwytny (jak np. w chorobach lizosomalnych) czy ostry, np. po szczepieniu, infekcji (jak w chorobach mitochondrialnych), urazie głowy czy gorączce (jak w leukodystrofii ze znikającą istotą białą [VWM – vanishing white matter]), a także analiza jej przebiegu (powoli postępujący czy falujący).11-13 Na właściwe rozpoznanie może również naprowadzić obecność objawów towarzyszących ze strony innych narządów i układów. Szczególnie wartościowe jest stwierdzenie oprócz cech hipoplazji lub zaniku nerwów wzrokowych dodatkowych zmian w narządzie wzroku (zaćma, retinopatia, zmętnienie rogówki).
Do ustalenia rozpoznania przyczynowego niezbędne są liczne badania laboratoryjne, w tym biochemiczne (np. ocena aktywności resztkowej enzymu, wykrycie nieprawidłowych metabolitów w płynach ustrojowych), elektrofizjologiczne, rzadziej – histo- i immunohistochemiczne hodowanych fibroblastów lub tkanek pobranych drogą biopsji.11-13 Biopsja, będąca badaniem inwazyjnym, zalecana jest coraz rzadziej, poza tym wymaga oceny w wyspecjalizowanych laboratoriach. Jak wspomniano wyżej, badania MR są podstawą w diagnostyce LD i gLE, jednak zawsze konieczna jest staranna analiza kliniczno-neuroobrazowa.11-13
Obecnie coraz częściej zaleca się badania molekularne w celu znalezienia patogennej mutacji w DNA. Koszty tych badań maleją, więc powinno się rozważyć ich wykonanie nawet na początku procedur diagnostycznych. Znalezienie mutacji nie tylko ostatecznie potwierdza przyczynę choroby, ale też umożliwia objęcie pacjenta i jego rodziny poradnictwem genetycznym. Pozwala również wdrożyć właściwe dla danej jednostki chorobowej postępowanie wspierające, rehabilitacyjne i zapobiegające powikłaniom, a co za tym idzie – zmniejszyć publiczne nakłady na lecznictwo.14,15
Leukodystrofie hipomielinizacyjne (HLD)
HLD są spowodowane trwałym brakiem mieliny, jej niedoborem i/lub nieprawidłową mielinizacją. Występują głównie u dzieci i młodzieży. Zależnie od patogenności mutacji mogą się ujawnić począwszy od okresu płodowego. Do dziś poznano ponad 14 genów leżących u podłoża HLD, ale wśród tego typu zaburzeń wciąż dominują choroby o nieustalonej etiologii.3,6 Leukodystrofie z dysmielinizacją (dysLD), czyli z niedostateczną i/lub nieprawidłową mielinizacją, wynikają najczęściej z aberracji chromosomowych.
Dysmielinizacja w obrazie MR stwierdzana jest również w łagodniejszych postaciach chorób zaliczanych do HLD, jak postać alleliczna choroby Pelizaeusa-Merzbachera (PMD – Pelizaeus-Merzbacher disease) – parapareza spastyczna typu 2 (SPG2 – spastic paraplegia type 2), a także w niektórych innych paraparezach spastycznych (m.in. SPG35).
W postaciach wrodzonych, które zaczynają się w okresie płodowym, obserwuje się najcięższy i niepomyślny przebieg. Chore dzieci są znacznie opóźnione psychoruchowo, na ogół bardzo wiotkie i – o ile nie rozwinęła się neuropatia obwodowa – mają zachowane odruchy. Niemal zawsze występuje oczopląs. W niektórych HLD pojawia się również padaczka. Szybko dochodzi do uspastycznienia i śmierci wskutek powikłań.
Postacie o późniejszym początku charakteryzują się nieswoistym, niekiedy nieomal stacjonarnym lub bardzo wolno postępującym przebiegiem, z przeważającym opóźnieniem ruchowym na skutek wiotkości. Wiotkość wynika głównie z uszkodzenia OUN, ale może być też efektem współistniejącej neuropatii obwodowej. Do zespołu piramidowego, a zwłaszcza spastyczności wynikającej z uszkodzenia istoty białej, dochodzi po wielu miesiącach, a nawet latach. Oczopląs jest nieomal stałym objawem, choć jego nasilenie może się zmniejszać z wiekiem. Inaczej niż w przypadku LDD prawie zawsze współistnieją objawy pozapiramidowe. W początkowym okresie przeważają dyskinezy, w miarę postępowania choroby pojawia się dystonia. Odruchy głębokie są zachowane, a dopiero z czasem i nie we wszystkich przypadkach stają się one wygórowane. W razie neuropatii mogą być osłabione. Objaw Babińskiego pojawia się nie zawsze i zazwyczaj po dłuższym okresie choroby. Rozwój umysłowy, emocjonalny i społeczny dzieci z HLD i dysLD jest względnie lepszy niż ruchowy, a niekiedy nawet prawidłowy. Stężenie białka w płynie mózgowo-rdzeniowym nie jest podwyższone, czasami bywa wręcz niskie.
Badanie MR ujawnia zmiany charakterystyczne dla hipomielinizacji/dysmielinizacji (najczęściej niewielka lub nierównomierna hiperintensywność istoty białej w sekwencji T2, zwłaszcza w T2-FLAIR, w stosunku do kory mózgu i zwojów podstawy).9,10 W niektórych HLD współistnieją zmiany w zwojach podstawy oraz zanik móżdżku, jak w HLD typu 6 uwarunkowanej mutacją genu TUBB4A. Ponadto na prawidłowe rozpoznanie może naprowadzić współistnienie zmian w innych narządach i układach, takich jak np. zaćma w HLD typu 4 uwarunkowana mutacją HSPD1 czy hipodoncja i/lub hipogonadyzm hipogonadotropowy w HLD typu 7 i 8.11-13
Choroba Pelizaeusa-Merzbachera (PMD)
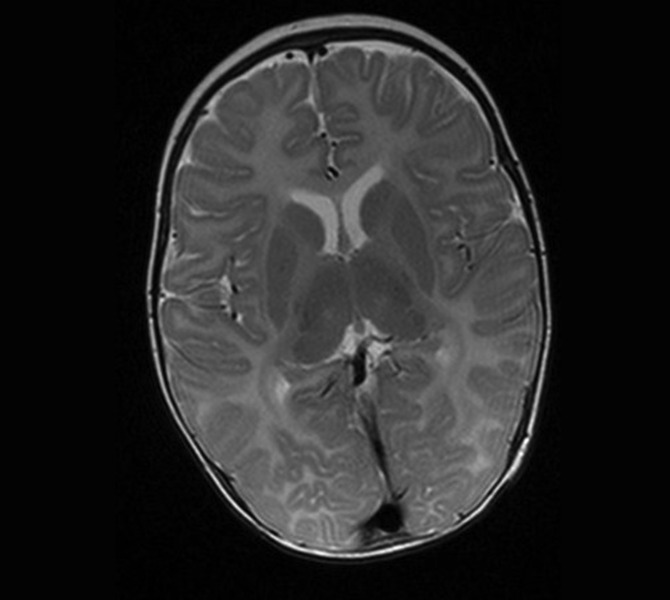
Rycina 1. Choroba Pelizaeusa-Merzbachera (HLD1). MR, sekwencja T2. Całkowita hipomielinizacja istoty białej mózgu łącznie z zajęciem torebek wewnętrznych. Chłopiec 2-letni z mieszanym zespołem pozapiramidowo-móżdżkowo-piramidowym, jeszcze bez spastyczności