Zespół CARASIL wywołany jest mutacją w genie HTRA1 na chromosomie 10q26.13, kodującym peptydazę serynową 1 HtrA, która hamuje przekazywanie sygnałów przez działanie na białka należące do rodziny transformujących czynników wzrostu β7. Częstość jego występowania nie jest znana. Większość chorych pochodzi z Japonii, pozostałe przypadki o powiązaniach rodzinnych opisano w Hiszpanii i Chinach6,7.
Początek choroby jest zmienny, a pierwszymi objawami są zwykle rozległe łysienie i zaburzenia chodu, które często zaczynają się przed ukończeniem 30 roku życia7. Napady ostrego bólu kręgosłupa w odcinku piersiowym lub lędźwiowym pojawiają się zazwyczaj między 20 a 45 r.ż. U połowy pacjentów występuje typowy udar niedokrwienny zatokowy, a u pozostałych następuje stopniowe pogorszenie czynności mózgu z postępującymi zaburzeniami poznawczymi, prowadzącymi do ciężkiego otępienia. Zaburzenia te zaczynają się ujawniać w wieku 30-40 lat. W stadiach zaawansowanych rozwijają się niestabilność emocjonalna, abulia i mutyzm akinetyczny. Chorzy są niezdolni do samodzielnej egzystencji mniej więcej po10 latach od początku zachorowania, ale mogą żyć 20-30 lat z chorobą6,7.
W rezonansie magnetycznym stwierdza się rozlane zmiany istoty białej o hiperintensywnych sygnałach oraz liczne udary lakunarne w okolicy jąder podkorowych, wzgórza i pnia mózgu7. Dodatkowo w obrazach kręgosłupa można uwidocznić liczne przepukliny jąder miażdżystych krążków międzykręgowych.
Histopatologicznie CARASIL przejawia się stwardnieniem małych przeszywających naczyń mózgu, bez GOM8. Rozpoznanie ustala się na podstawie obecności charakterystycznych objawów klinicznych oraz wyników obrazowania rezonansu magnetycznego. Diagnozę potwierdza badanie genetyczne wykazujące mutację w HTRA1.
Zespół CARASAL
Zespół CARASAL (katepsynozależna arteriopatia z udarami i leukoencefalopatią; cathepsin A-related arteriopathy with strokes and leukoencephalopathy) jest chorobą małych naczyń spowodowaną mutacją genu CTSA na chromosomie 20q13.12, kodującego katepsynę A9. Najczęstszymi objawami są migreny z aurą oraz niedokrwienne udary mózgu. Mogą występować również krwotoki podpajęczynówkowe. Średni wiek zachorowania to 35 lat, a narastające otępienie jest bardzo nasilone już w wieku około 40 lat. Choroba jest bardzo rzadka (dotychczas opisano jedynie kilka przypadków na świecie)9.
O rozpoznaniu CARASAL należy myśleć u młodych pacjentów z chorobą małych naczyń oraz nasilonymi zmianami w istocie białej w wywiadzie rodzinnym, przy braku mutacji genów NOTCH3, HTRA1 i COL4A1/A29. W takich przypadkach rozstrzygające jest badanie genetyczne w kierunku mutacji genu CTSA.
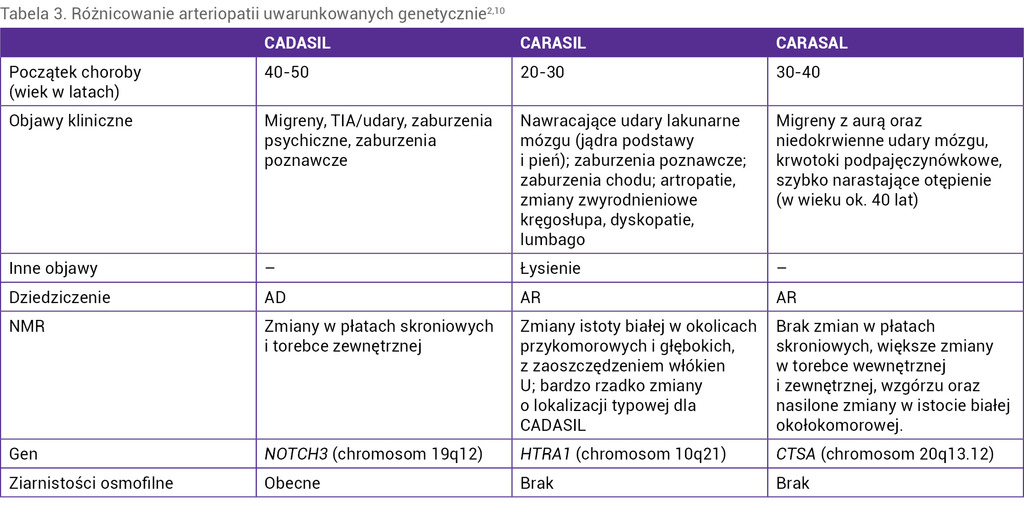
Tabela 3. Różnicowanie arteriopatii uwarunkowanych genetycznie2,10
Różnicowanie arteriopatii uwarunkowanych genetycznie przedstawiono w tabeli 3.
Choroba Fabry’ego
Choroba Fabry’ego jest lizosomalną chorobą spichrzeniową dziedziczoną z chromosomem X. Objawy występujące u kobiet związane są z przypadkową inaktywacją chromosomu X10. Przyczyną choroby jest mutacja genu GLA (Xq22) kodującego α-galaktozydazę A (α-Gal A; triheksozydazę ceramidową). Niedobór enzymu powoduje upośledzenie metabolizmu globotriaosylceramidu i jego gromadzenie w lizosomach komórek naczyń, komórek zwojów nerwowych, nerkach, sercu, gałkach ocznych i innych narządach12. Dochodzi do uszkodzenia komórek śródbłonka naczyń, mięśnia sercowego, komórek nerwowych i komórek układu autonomicznego. Częstość występowania choroby szacuje się na 1/117 000 żywych urodzeń10.
Choroba ma zróżnicowany obraz kliniczny. Najczęstszymi objawami są akroparestezje, zmiany skórne (angiokeratoma), białkomocz i niewydolność nerek oraz zaburzenia neurologiczne (udary mózgu, TIA)11. Objawy pojawiają się zazwyczaj między 6 a 9 rokiem życia. U pacjentów z łagodną postacią choroby Fabry’ego i rezydualną aktywnością α-Gal A objawy pojawiają się w wieku dorosłym i zazwyczaj ograniczone są do chorób serca i nerek10-12.
Przyjęto podział na trzy postacie choroby10:
- postać klasyczna – cechuje się wczesnym początkiem (w wieku przedszkolnym), aktywnością enzymu <1% i występowaniem wszystkich objawów klinicznych
- postać nerkowa – rozpoczyna się około 25 r.ż., aktywność enzymu wynosi >1%, dominują objawy niewydolności nerek i kardiomiopatia, nie występuje jednak uszkodzenie OUN
- postać sercowa – rozpoczyna się około 40 r.ż., aktywność enzymu wynosi >1%, dominują objawy kardiomiopatii.
W pierwszej dekadzie życia choroba objawia się bólami mięśniowymi i parestezjami kończyn, w drugiej – zaburzeniami potliwości, bólami brzucha i biegunkami. W trzeciej dekadzie często dołącza się niewydolność nerek z proteinurią, lipidurią i krwiomoczem, a w czwartej – kardiomiopatia, przerost lewego przedsionka, uszkodzenia zastawek, zaburzenia przewodzenia. Objawy neurologiczne ujawniają się w piątej dekadzie życia11. Udary mózgu i przemijające niedokrwienia mózgu obserwowane są 12 razy częściej niż w populacji ogólnej (średnia wieku 25-44 lata)10.Charakterystyczna jest predylekcja do zajęcia naczyń tylnego kręgu. Typowe są dolichoektazje (odcinkowe poszerzenia i kręty przebieg) tętnic kręgowych i podstawnej. W późniejszym czasie dołączają się zaburzenia poznawcze i pozapiramidowe, neuropatia aksonalna, szumy uszne i utrata słuchu10-12.
Do charakterystycznych objawów należą również gorączka nieznanego pochodzenia, zaburzenia wydzielania potu (anhydroza), angiokeratoma (ciemnoczerwone, rogowaciejące naczyniaki na skórze bioder, ud, krocza) oraz neuropatyczne bóle kończyn, szczególnie dłoni i stóp, wymagające podawania opioidów. Mogą wystąpić zaburzenia adaptacyjne i depresyjne. Najczęstszymi objawami neurologicznymi są niedowłady połowicze i objawy z tylnego kręgu krążenia mózgowego10.
U pacjenta, u którego objawy sugerują chorobę Fabry’ego, należy wykonać badanie stężenia α-Gal A w surowicy krwi i leukocytach. Możliwe są badania prenatalne oraz u członków rodziny chorego pozostających bez objawów, umożliwiające wczesne rozpoczęcie leczenia13.
Obraz MR mózgowia wykazuje cechy uszkodzenia małych naczyń pod postacią rozsianych hiperintensywnych ognisk w istocie szarej i białej płatów czołowych i ciemieniowych w obrazach T2 i FLAIR15. Niemal patognomoniczny jest objaw poduszki – obustronnie hiperintensywny sygnał poduszki (złogi wapnia) w tylnej części wzgórza na obrazach T1-zależnych14. Występuje u około 23% chorych w trzeciej dekadzie życia. Za chorobą Fabry’ego przemawiać może również poszerzenie i kręty przebieg tętnic kręgowych i tętnicy podstawnej w badaniu angio-MR. Inne zmiany są mniej swoiste i obejmują ogniska porencefaliczne po przebytych udarach oraz zmiany istoty białej – rozsiane ogniska naczyniopochodne i rozlane zmiany okołokomorowe14.
Należy również przeprowadzić badania czynności nerek, EKG, badania echokardiograficzne, okulistyczne w lampie szczelinowej oraz dermatologiczne13.