Minisympozjum – endokrynologia
Wrodzony przerost nadnerczy u dzieci i młodzieży – diagnostyka i leczenie
Maria Ginalska-Malinowska
Słowa kluczowe
wrodzony przerost nadnerczy, hormony steroidowe
Wprowadzenie
Kora nadnerczy jest miejscem powstawania 3 rodzajów hormonów steroidowych: mineralokortykosteroidów, glikokortykosteroidów oraz androgenów. Wszystkie te hormony powstają z cholesterolu, a poszczególne etapy syntezy przebiegają dzięki obecności wielu różnych enzymów – substancji białkowych działających na określone substraty i powodujących powstanie kolejnych produktów steroidogenezy (ryc. 1). Najważniejszymi hormonami wytwarzanymi w tym procesie u człowieka są: aldosteron (mineralokortykosteroid), kortyzol (glikokortykosteroid) oraz dehydroepiandrosteron, androstendion i testosteron (androgeny).
Zmniejszenie aktywności któregoś z enzymów koniecznych do przebiegu prawidłowej steroidogenezy w korze nadnerczy prowadzi do zmian w przebiegu tego procesu, co skutkuje niedostatecznym wytwarzaniem niektórych hormonów i nadmiarem innych. Zaburzenia steroidogenezy prowadzą najczęściej do niedostatecznego wytwarzania najważniejszego hormonu kory nadnerczy – kortyzolu. Przewlekły niedobór kortyzolu powoduje stałe pobudzenie układu podwzgórzowo-przysadkowego do zwiększonego wydzielania kortykoliberyny (CRH) oraz kortykotropiny (ACTH), działających stymulująco na korę nadnerczy i powodujących jej przerost. Dlatego chorobę związaną z zaburzeniami syntezy steroidów w korze nadnerczy na skutek braku właściwego działania enzymów steroidogenezy nazywa się wrodzonym przerostem nadnerczy (WPN) (congenital adrenal hyperplasia, CAH). Jest to szczególna forma przewlekłej niedoczynności kory nadnerczy, w której niedobór kortyzolu współistnieje z nadmiarem i/lub niedoborem innych hormonów kory nadnerczy. Nieprawidłowości te mają charakter wrodzony, zależą od mutacji w genach kodujących dany enzym i dziedziczą się autosomalnie recesywnie.1 Oznacza to, że objawy kliniczne ujawniają się u homozygot, zaś osoby z mutacją genu na jednym allelu są bezobjawowymi nosicielami choroby.
Epidemiologia i objawy kliniczne
Objawy kliniczne WPN są zróżnicowane i zależą od tego, na jakim etapie steroidogenezy i w jakim nasileniu występuje ograniczenie aktywności enzymu. Najczęściej, bo aż w 90-95% przypadków WPN stwierdza się niedobór enzymu 21-hydroksylazy.1,2 Zaburzenia steroidogenezy związane z niedoborami innych enzymów (oznaczonych symbolami P450scc, P450c17, 3β-HSD, P450c11β, P450c11AS na ryc. 1) rozpoznaje się niezwykle rzadko, dlatego dalsza część artykułu będzie dotyczyć tylko WPN zależnego od niedoboru 21-hydroksylazy (P450c21).
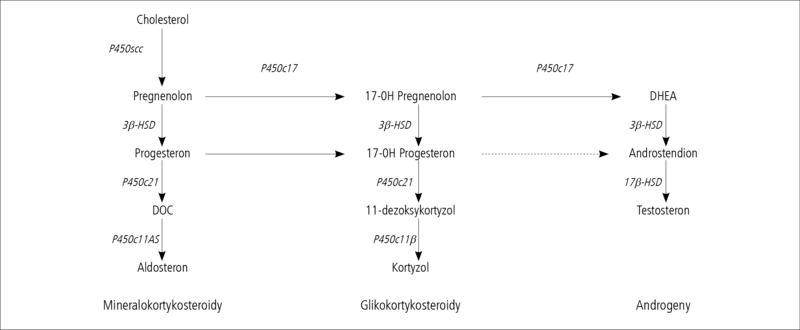
Rycina 1. Schemat steroiodgenezy w korze nadnerczy.
W nadnerczach osoby chorej na ten typ WPN dochodzi do nagromadzenia 17-OH progesteronu (17-OHP) – związku, którego dalsza przemiana do kortyzolu nie jest możliwa z powodu niedostatecznej aktywności 21-hydroksylazy. Znaczące zwiększenie ilości 17-OHP i brak możliwości dalszych jego przemian we właściwym kierunku prowadzi do „przekierowania” steroidogenezy w stronę androgenów, w syntezie których 21-hydroksylaza w ogóle nie bierze udziału (ryc. 2). Dlatego to właśnie objawy hiperandrogenizacji nasuwają zwykle podejrzenie WPN.
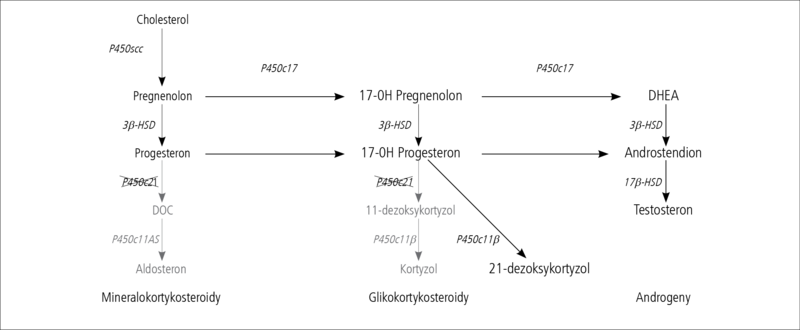
Rycina 2. Schemat steroiodogenezy w korze nadnerczy przy niedoborze 21-hydroksylazy.
Wyróżnia się kilka postaci klinicznych choroby: a) klasyczną z utratą soli (salt wasting,SW), ze znacznym niedoborem kortyzolu i aldosteronu oraz nadmiarem androgenów – z klinicznymi objawami choroby od pierwszych dni/tygodni po urodzeniu, b) klasyczną bez utraty soli (simple virilizing, SV), z istotnym niedoborem kortyzolu i nadmiarem androgenów, bez ograniczenia syntezy aldosteronu, rozpoznawaną zwykle w 1 roku życia lub we wczesnym dzieciństwie, głównie z powodu szybko narastających objawów androgenizacji, c) nieklasyczną (non classic, NC) z łagodnym nadmiarem androgenów, bez niedoboru mineralokortykosteroidów oraz bez klinicznych objawów hipokortyzolemii.
Częstość występowania postaci klasycznej ocenia się na 1:10 000-1:15 000 noworodków, natomiast nieklasycznej na 1:1000 osób, z częstością wyraźnie większą (1:200-1:400) wśród mieszkańców basenu Morza Śródziemnego oraz w populacji Żydów aszkenazyjskich.1
Choroba dziedziczy się autosomalnie recesywnie, dotyczy więc zarówno dziewcząt, jak i chłopców. Objawy ogólne, zwane zespołem utraty soli, pojawiają się u większości (75%) chorych noworodków obu płci, stwarzając poważne zagrożenie życia. Są spowodowane znacznym ograniczeniem wydzielania aldosteronu i kortyzolu. Występują zwykle nie wcześniej niż w 6-8 dobie życia dziecka, czyli dopiero wówczas, gdy dochodzi do fizjologicznego zaniku warstwy płodowej kory nadnerczy (stanowiącej po urodzeniu 85% masy gruczołu). Objawy utraty soli to: brak przyrostu masy ciała, cechy odwodnienia, brak łaknienia, wymioty, senność, obniżenie ciśnienia tętniczego, pogarszanie się stanu ogólnego. Według mojej obserwacji dość stałym objawem zespołu utraty soli w pierwszych dniach życia jest większe niż fizjologiczne zmniejszenie masy ciała noworodka i brak odpowiednio szybkiego osiągnięcia ponownie masy urodzeniowej.
Jak wspomniano, niedobór 21-hydroksylazy u chorych z klasyczną postacią WPN (zarówno z utratą, jak i bez utraty soli) przebiega ze znacznym nadmiarem androgenów nadnerczowych. Hiperandrogenizm rozpoczyna się już w okresie płodowym, co jest szczególnie niekorzystne dla płodów żeńskich, gdyż powoduje nieodwracalną maskulinizację zewnętrznych narządów płciowych w czasie ich embriogenezy, która zachodzi od 9 do 15 tygodnia życia płodowego.1 Może to powodować trudności w określeniu płci dziecka po urodzeniu, nierzadko jest nawet przyczyną niewłaściwego określenia płci jako męska u chorej dziewczynki (WPN z całkowitym odwróceniem płci). Chłopcy z klasyczną postacią WPN rodzą się z prawidłowymi narządami płciowymi, co powoduje, że nawet mimo typowych objawów zespołu utraty soli rozpoznanie choroby i rozpoczęcie odpowiedniego leczenia jest często opóźnione.3,4 Podejrzewa się u takich noworodków choroby zapalne przewodu pokarmowego, zwężenie odźwiernika lub inne przyczyny niedoboru masy ciała.
Klasyczna postać WPN bez utraty soli u dziewczynek rozpoznawana jest zwykle w wieku niemowlęcym lub wkrótce potem ze względu na widoczną maskulinizację narządów płciowych. U chłopców objawy narastającej androgenizacji (przyspieszenie tempa wzrastania ze zbyt szybkim awansowaniem wieku kostnego, objawy przedwczesnego dojrzewania płciowego) są diagnozowane przeważnie dopiero w wieku przedszkolnym. Dość częstym objawem WPN u chłopców w okresie pokwitania są zmiany guzowate w jądrach. Zmiany te są najczęściej ogniskami tkanki nadnerczowej (testicular adrenal rest tissue, TART).5,6 Histologicznie przypominają skupiska komórek Leydiga, dlatego mogą być mylnie rozpoznawane jako leydigioma. TART stwierdza się częściej u chorych z WPN z cechami klinicznego i biochemicznego niedostatecznego wyhamowania ACTH, nawet u dzieci, mimo leczenia.6
Nieklasyczna postać WPN, z częściowym tylko zmniejszeniem aktywności enzymu, klinicznie objawia się łagodną androgenizacją ujawniającą się dopiero ok. 8-9 roku życia lub w wieku dojrzewania. Mimo że dotyczy obu płci, rozpoznawana jest głównie u dziewcząt ze względu na objawy androgenizacji: przedwczesne pubarche, hirsutyzm, nasilony trądzik lub jedynie zaburzenia miesiączkowania. Obraz kliniczny nieklasycznej postaci WPN u dziewcząt przypomina często objawy zespołu policystycznych jajników, choć niewykluczone jest współistnienie obu tych chorób jako następstwo przewlekłego hiperandrogenizmu w przebiegu WPN. Łagodny nadmiar androgenów w nieklasycznej postaci WPN, szczególnie bez zaburzeń miesiączkowania, nie zawsze wymaga leczenia farmakologicznego.3,4
U chorych z klasycznym WPN częściej niż w ogólnej populacji stwierdza się otyłość i cechy insulinooporności. Jest to spowodowane istniejącą w WPN niedoczynnością rdzenia nadnerczy, a szczególnie zaburzeniem syntezy adrenaliny. Hipokortyzolemia, hiperandrogenizm i niedobór adrenaliny prowadzą nieuchronnie do wielu wtórnych zmian metabolicznych, nasilających główne objawy choroby.7 Zaburzenia metaboliczne pogłębiają się szczególnie w okresie pokwitania, co dodatkowo komplikuje zasadnicze leczenie.
DO ZAPAMIĘTANIA
• WPN wynikający z niedoboru 21-hydroksylazy jest najczęstszą endokrynologiczną przyczyną niewydolności kory nadnerczy u niemowląt oraz główną przyczyną przedwczesnego dojrzewania płciowego u chłopców w wieku przedszkolnym.
• Nieprawidłowości w wyglądzie narządów płciowych u noworodka powinny nasuwać podejrzenie WPN. W związku z koniecznością wykonania specjalistycznych badań diagnostycznych niezbędnych do ustalenia rozpoznania w ośrodku referencyjnym pourodzeniowa rejestracja takiego noworodka (jak każdego z zaburzeniami różnicowania płci) powinna być odroczona.
• Zespół utraty soli rozwija się najczęściej dopiero w końcu 1 tygodnia życia. Dlatego brak typowych zaburzeń w jonogramie w pierwszej/pierwszych dobach po urodzeniu nie wyklucza WPN u noworodka.