Ortopedia pediatryczna
Obraz kliniczny i wybrane elementy fizjoterapii dzieci z wrodzoną łamliwością kości, czyli co każdy pediatra wiedzieć powinien
Elżbieta Jelonek,1 Krzysztof Graff2
Wprowadzenie
Moja „przygoda” z wrodzoną łamliwością kości rozpoczęła się ponad ćwierć wieku temu, kiedy jako młoda lekarka, tuż po uzyskaniu specjalizacji z pediatrii, rozpoczęłam pracę na oddziale rehabilitacji Centrum Zdrowia Dziecka. Moje wyobrażenia o tym schorzeniu były wręcz symboliczne – w czasie, gdy studiowałam, Sillence jeszcze nie ogłosił światu podziału tego schorzenia na poszczególne typy, a wiedza książkowa ograniczała się do kilku zdań. Nie byłam więc świadoma, z czym przyjdzie mi się zmierzyć. Poszukiwania informacji o tej chorobie były jak droga przez mękę, a młodsi koledzy nie mają wyobrażenia o tym, z jakim trudem w tamtych siermiężnych latach zdobywało się informacje medyczne. Dziś stukamy w klawiaturę, siedząc w domu, w pracy, w parku na ławeczce, jadąc autobusem do pracy i cała wiedza pcha się do nas z każdego zakątka świata. Najwięcej jednak doświadczenia zdobyłam dzięki codziennej pracy przy rehabilitacji dzieci z tym schorzeniem. Specjalizacja z rehabilitacji medycznej to nie tylko wiedza ściśle lekarska. Poza merytorycznymi informacjami o istocie tego schorzenia (a także każdego, z którym specjalista z zakresu rehabilitacji ma na co dzień do czynienia) jest to ścisła współpraca z fizjoterapeutą, który jest kreatorem sukcesu terapeutycznego. Bez dobrego terapeuty lekarz nie może oczekiwać dobrych, znaczących efektów, bo wszystkie znane i dostępne metody leczenia muszą być nierozerwalnie skoordynowane z mądrą, czyli nowoczesną, wiedzą z zakresu fizjoterapii. Miałam szczęście współpracować z terapeutami rozumiejącymi zagadnienie i w miarę, jak poszerzała się wiedza medyczna w tej dziedzinie, oni też modyfikowali swoje umiejętności. To właśnie praca w Klinice Rehabilitacji uświadomiła mi, że kolejną moją specjalizacją będzie rehabilitacja medyczna. Przerwałam rozpoczęty II stopień z zakresu pediatrii i od ponad 20 lat moją codziennością jest rehabilitacja.
Rozwój wiedzy medycznej, postęp w technikach operacyjnych zmienił oblicze tej ciężkiej choroby, jaką jest wrodzona łamliwość kości. Ale, jak mówi łacińska sentencja, durante causa, durant effectus (dopóki działa przyczyna, trwa i skutek), a osteogenesis imperfecta to choroba uwarunkowana genetycznie, sporo wody w Wiśle upłynie, zanim będziemy mogli takie genetyczne wybryki leczyć naprawdę, a nie tylko próbować coraz skuteczniej oszukiwać biologię.
Wstęp
Wrodzona łamliwość kości (osteogenesis imperfecta, OI) jest rzadkim schorzeniem tkanki łącznej uwarunkowanym genetycznie, o niezwykle dużym zróżnicowaniu ekspresji objawów klinicznych, heterozygotycznym pod względem dziedziczenia.1-3 Wspólne dla wszystkich postaci jest zaburzenie budowy kolagenu typu I, będącego głównym białkiem strukturalnym wchodzącym w skład macierzy kostnej, ścian naczyń krwionośnych, skóry i ścięgien.4 Zanim kolagen osiągnie ostateczną formę, poddawany jest wieloetapowej obróbce zarówno wewnątrz-, jak i zewnątrzkomórkowej.5-9 Łańcuch kolagenu składa się z trzech łańcuchów polipeptydowych połączonych i splecionych w jeden warkocz. Dwa z nich, tzw. łańcuchy α1, są identycznej budowy, a trzeci łańcuch α2 różni się składem aminokwasów. Istotną cechą kolagenu jest powtarzająca się sekwencja aminokwasów o wzorze X-Y-Z, gdzie na miejscu Y i Z jest prolina lub hydroksyprolina, a na miejscu X glicyna.10-12 Zamiana glicyny na inny aminokwas skutkuje określonym obrazem fenotypowym.10 Najczęściej wykrywane są zamiany glicyny na serynę lub tryptofan.4,12 Defekty kolagenu wynikają z błędów na różnych etapach jego powstawania i w większości są rezultatem mutacji genów strukturalnych lub genów regulujących sekwencje aminokwasów w łańcuchach białkowych.5,13,14
Historia

Tabela. Charakterystyka kliniczna choroby oparta na klasyfikacjo Sillence’a
Wrodzona łamliwość kości występowała już w starożytności, o czym świadczą znaleziska archeologiczne. W British Museum znajdują się szczątki osoby z wrodzoną łamliwością kości, które datują się na 1000 r. p.n.e.15 Od ponad 200 lat opisywano to schorzenie pod różnymi nazwami. Jako pierwszy chorobę opisał Nicolas Malebrache w 1678 r., a Szwed Jakub Ekman użył określenia osteomalacja congenita. W 1826 r. Sartorius użył nazwy rachitis congenita, a Lobstein w 1833 r. opisał ją jako osteopsathyrosis idiopatica. Obecnie stosowaną nazwę osteogenesis imperfecta jako pierwszy podał Holender Vrolik w 1849 r.16 W 1906 r. Loosem zaproponował podział wrodzonej łamliwości kości na dwie grupy: postać ciężką, zwaną inaczej chorobą Vrolika, oraz postać późną, zwaną chorobą Lobsteina. Seedorf w 1949 r. wyróżnił dwa podtypy OI – osteogenesis tarda gratis, jeśli choroba ujawnia się w pierwszym roku życia, oraz osteogenesis tarda levis, jeśli do pierwszych złamań dochodzi w dzieciństwie lub później.16-18 Stopień nasilenia zmian w narządzie ruchu i tkankach zawierających kolagen oraz typ dziedziczenia stanowił podstawę do przyjęcia podziału OI, zgodnie z wytycznymi ustalonymi w 1986 r. na Międzynarodowym Kongresie Genetyki Człowieka, na 4 główne typy z podtypami wg Sillence’a (tabela).19-21 Przedstawiona klasyfikacja OI przyjmowała jako kryterium podziału sposób dziedziczenia, obraz radiologiczny, częstość występowania oraz różnice w obrazie klinicznym.7,13,22 W 2004 r. w czasopiśmie Lancet opublikowano wyniki seminarium dotyczącego OI, w których rozbudowano dotychczasową klasyfikację, wyodrębniając typy I-VII,23-25 a w 2007 r. zaproponowano, aby dodać typ VIII.26
Diagnostyka kliniczna
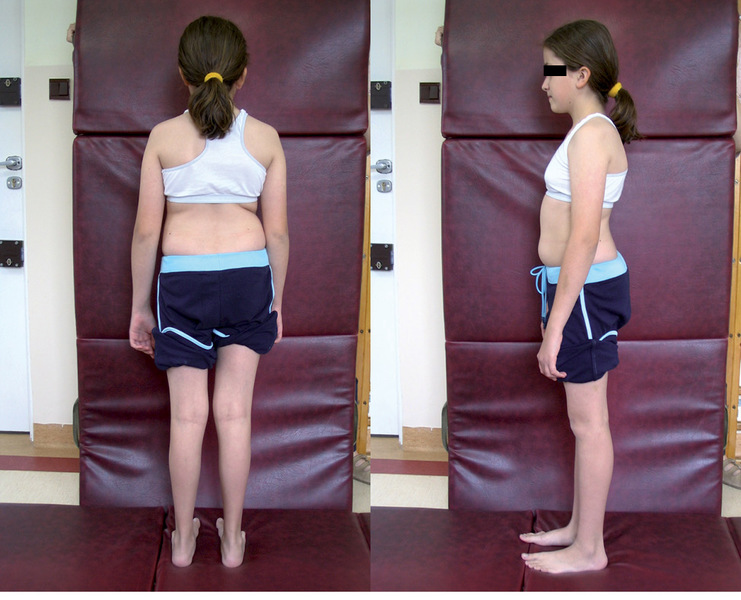
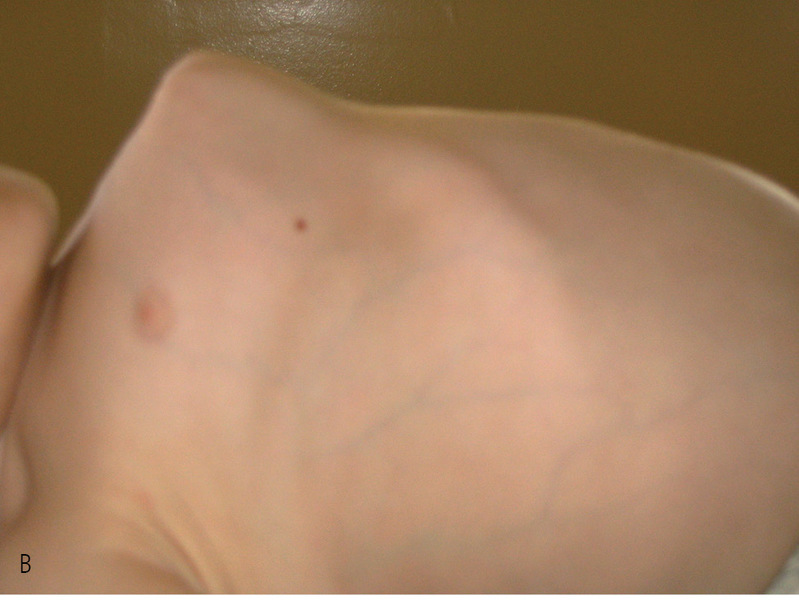
Rycina 1. Dziewczynka z typem I OI.
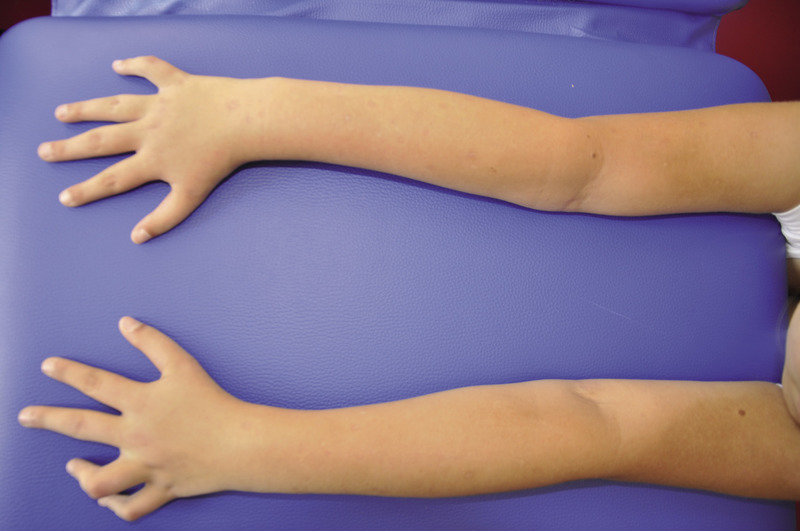
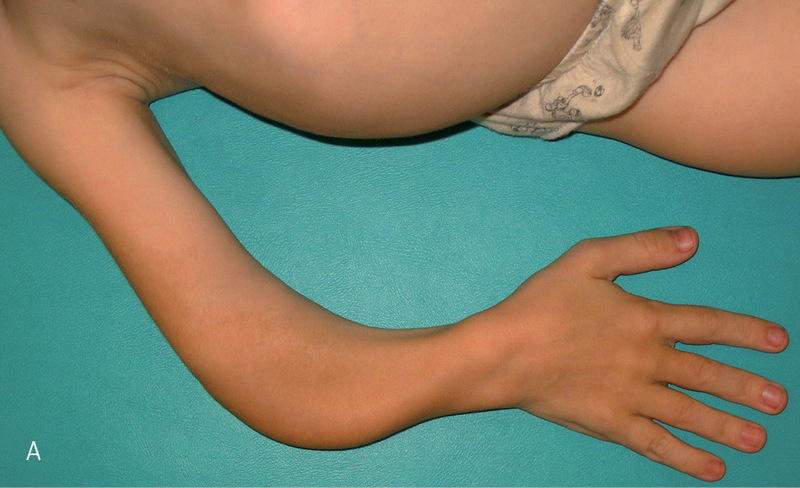
Rycina 2. Typ I A – deformacje kończyn górnych, charakterystyczne przeprosty w stawach kolanowych, nieznaczna koślawość w stawach kolanowych oraz stopy płasko-koślawe.
W ciężkich postaciach schorzenia dokładne badanie przedmiotowe i prawidłowo przeprowadzony wywiad powinny nasunąć nawet początkującemu klinicyście rozpoznanie typu strassen diagnose (bez wykonania dodatkowej diagnostyki). W typach o łagodniejszym przebiegu konieczne jest wykonanie badań radiologicznych, densytometrycznych oraz gospodarki wapniowo-fosforanowej (Ca-P).
Typ I wrodzonej łamliwości kości
Typ I (ryc. 1 i 2) z podtypami I A i I B jest najłagodniejszą i najczęściej spotykaną postacią tej choroby. Według różnych autorów szacuje się, że choroba występuje z częstością 1:15000-1:20000 urodzeń.13 Dzieci z tym typem charakteryzują się uogólnioną dystonią mięśniową. Poruszają się samodzielnie po różnym terenie. Ich wzrost nie odbiega znacząco od normy rówieśniczej dzieci zdrowych.27 Deformacje układu kostnego są zwykle niewielkie i dotyczą najczęściej jednego z odcinków kości długich. Wśród 75% chorych stwierdza się nadmierną ruchomość stawów z tendencją do podwichnięć i zwichnięć. Chorzy z typem I A mają nieznaczny niedobór wzrostu, który ujawnia się dopiero w kilka lat po urodzeniu. Chorych z typem I B charakteryzuje niskorosłość i większa tendencja do złamań po urodzeniu.
Typ II wrodzonej łamliwości kości
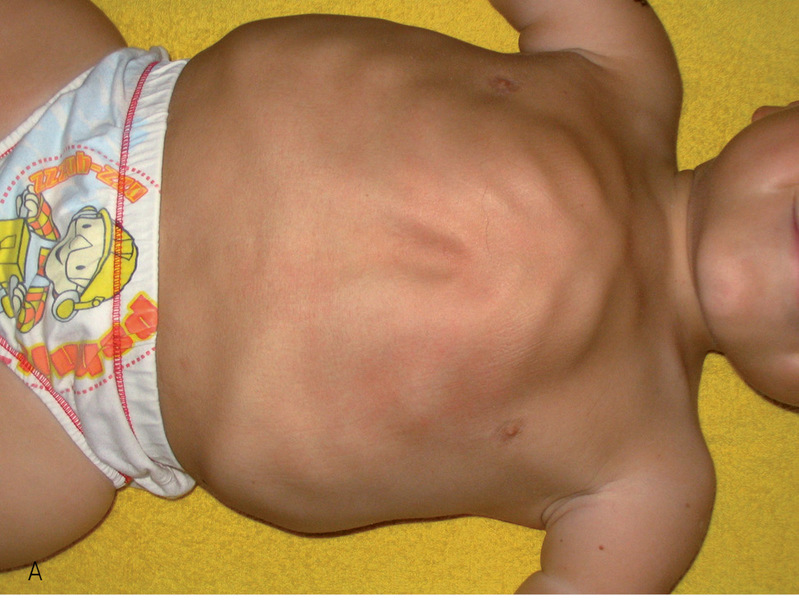
Rycina 3. Typowe deformacje klatki piersiowej i żeber u dzieci z typem III OI.
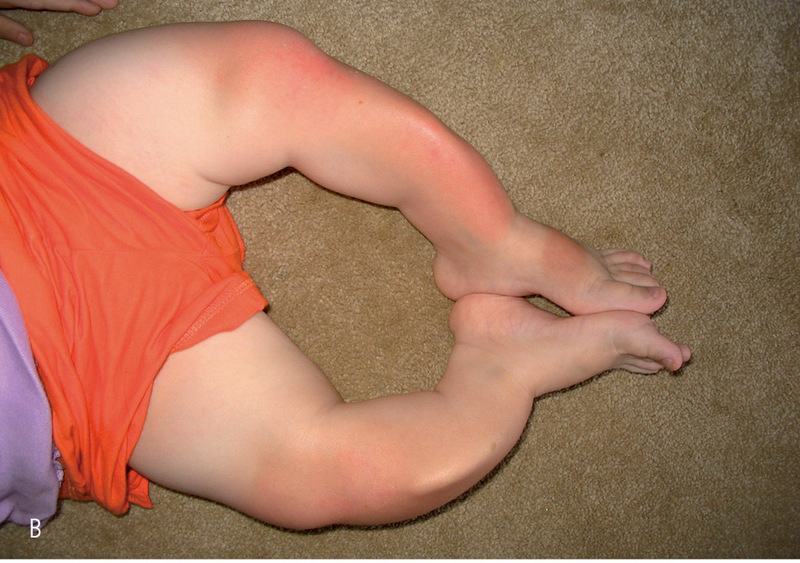
Rycina 4. Deformacje kończyn. (A) Deformacja kończyn górnych, (B) dolnych (szablaste wygięcia).

Rycina 5. Objawy hipermobilności stawowej u chorych z typem III OI.

Rycina 6. Deformacja miednicy i kręgosłupa II/III° u dziewczynki z III typem OI.
Typ II jest najcięższą, letalną postacią choroby. Rozpoznawany jest z częstością 1:60000 urodzeń8,28 i częściej dotyczy dziewczynek. Około połowa dzieci rodzi się martwa, a w ciągu pierwszej doby z powodu niewydolności krążeniowo-oddechowej lub krwotoku wewnątrzczaszkowego umiera ok. 60% dzieci. Przeżycie ponad 1 roku należy do rzadkości. Dzieci z tym typem są zwykle urodzone przedwcześnie, z niedoborem długości i masy urodzeniowej. Rodzą się z licznymi złamaniami i deformacjami powstającymi wewnątrzłonowo. Ten typ choroby dziedziczony jest autosomalnie recesywnie i w większości przypadków jest wynikiem nowej mutacji.