


Spis treści
Słowa kluczowe
3-metylokrotonyloglicynuria, spektrometria mas, deficyt karboksylazy 3-metylokrotonylo-koenzymu A
Wprowadzenie
Dzięki badaniom przesiewowym noworodków metodą tandemowej spektrometrii mas, ostatnio powszechnie wykorzystywanym w diagnostyce rzadkich wrodzonych wad metabolizmu, identyfikowane są liczne przypadki chorób o nieudowodnionym dotychczas znaczeniu klinicznym (w tym 3-metylokrotonyloglicynuria). Często u zidentyfikowanych noworodków nie występują żadne niepokojące objawy
w dłuższym okresie obserwacji. W związku z tym pojawiają się wątpliwości co do konieczności wprowadzenia interwencji medycznej w postaci: zmiany diety dziecka (ograniczenie białka naturalnego, szczególnie leucyny), farmakoterapii (suplementacja L-karnityny), stałych zaleceń unikania przedłużonego głodzenia i natychmiastowego działania w sytuacjach wzmożonego katabolizmu (zagrożenie dekompensacją metaboliczną). Niezależnie od powyższych zaleceń większość klinicystów potwierdza potrzebę monitorowania przebiegu choroby mimo pojawiających się zarzutów dotyczących medykalizacji. 1
Co to jest 3-metylokrotonyloglicynuria (deficyt MCC)?
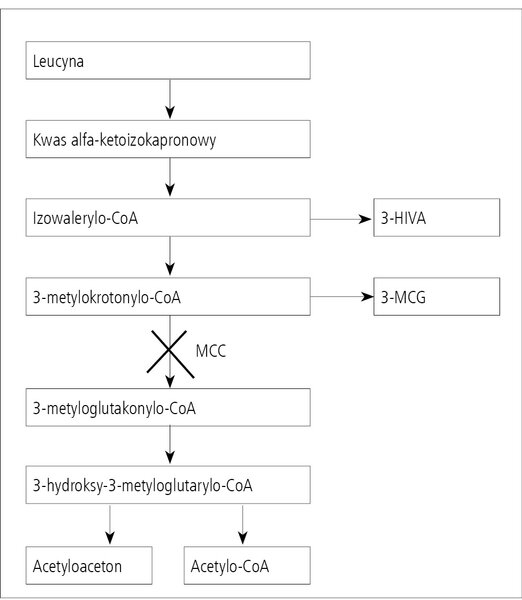
Rycina 1. Schemat przemiany leucyny z zaznaczeniem miejsca deficytu MCC.
Rozpoznawanie i fenotyp deficytu MCC przed erą badań przesiewowych noworodków
Jeszcze do niedawna deficyt MCC uznawany był za jedną z klasycznych acydurii organicznych z grupy rzadkich wrodzonych wad metabolizmu pośredniego aminokwasów rozgałęzionych, które w wyniku bloku enzymatycznego przebiegają z gromadzeniem kwasów karboksylowych, co powoduje objawy przypominające ostre zatrucie, czyli tzw. zespół intoksykacji. Do obrazu klinicznego deficytu MCC należały głównie nawracające epizody dekompensacji klinicznej (związanej ze stanami wzmożonego katabolizmu lub nadmiernego obciążenia białkiem) oraz dysfunkcja wątroby w postaci zespołu Reye’opodobnego, hipoglikemia i objawy neurologiczne (hipotonia, drgawki, dystonia, epizody udaropodobne, leukodystrofia, opóźnienie rozwoju psychoruchowego). 6, 7, 8 Charakterystyczny dla choroby jest też szczególny zapach wyczuwalny od osób chorych, przypominający zapach moczu kocura. Po ustaleniu rozpoznania postępowanie polegało na stosowaniu diety z ograniczeniem białka naturalnego wykorzystującej środki spożywcze specjalnego przeznaczenia medycznego niezawierające leucyny, z zaleceniem zapewnienia podaży kalorycznej w stanach wzmożonego katabolizmu oraz suplementacji karnityny w przypadku stwierdzenia jej niedoboru.
Rozpoznawanie i fenotyp deficytu MCC obecnie
W ostatnim czasie badania przesiewowe noworodków metodą tandemowej spektrometrii mas (tandem MS, MS/MS) ujawniły inne od dotychczas znanego oblicze tej wrodzonej wady metabolizmu. Okazało się, że zwiększone (wykraczające poza zakres wartości referencyjnych) stężenie C5OH wykrywane w suchej kropli krwi stanowi bardzo często stwierdzane odchylenie w badaniu przesiewowym; według Baumgartnera i wsp. u 1 na 50 000 noworodków, 9 według Arnold i wsp. u 1 na 36 000 noworodków. 1 Jednocześnie zakres obserwowanych objawów deficytu MCC znacznie się poszerzył – od ciężkich postaci noworodkowych do braku objawów nawet do wieku dorosłego. Różnorodność obrazu klinicznego (fenotypu) może wystąpić nawet w obrębie tej samej rodziny, mającej zatem ten sam genotyp. W pracy Grünert i wsp. wśród 8 pacjentów zdiagnozowanych jako cierpiących na deficyt MCC w ramach diagnostyki rodzinnej u 3 nie występowały objawy, u kolejnych 3 objawy były obecne, na temat pozostałych 2 brakuje danych. 10 Wśród objawów klinicznych stwierdzano: opóźnienie rozwoju psychoruchowego, opóźnienie rozwoju mowy, obniżenie napięcia mięśniowego, nadpobudliwość ruchową i niechęć do jedzenia mięsa. W ramach diagnostyki rodzinnej czteroletniego chłopca z deficytem MCC (hipotonią i miernym opóźnieniem rozwoju ruchowego) wykryto to zaburzenie u jego 17-letniego brata bez objawów, a w przypadku chłopca z drgawkami atonicznymi i kwasicą metaboliczną stwierdzonymi w wieku niemowlęcym (zdiagnozowany w ramach skriningu selektywnego) – u 3-letniego brata z opóźnieniem rozwoju mowy i makrocefalią potwierdzono deficyt MCC.
Visser i wsp. opisali występowanie deficytu MCC u 7-tygodniowej dziewczynki z kardiomiopatią rozstrzeniową, jej 10-letniego brata z niepełnosprawnością intelektualną, ale też u ich zdrowego ojca. Rozpoznanie u dwóch ostatnich ustalono w ramach diagnostyki rodzinnej po wykryciu nieprawidłowości u niemowlęcia. W konkluzji jednak autorzy sugerowali, że w tej rodzinie nie można wykluczyć odmiennego genotypu u poszczególnych członków rodziny. 11
Ustalenie ostatecznego rozpoznania dodatkowo utrudnia możliwość wystąpienia odchylenia pochodzenia odmatczynego wykrywanego u noworodka metodą MS/MS. Wiadomo bowiem, że matka z deficytrm MCC (zwykle o nim niewiedząca) karmiąca piersią przekazuje dziecku metabolity, które są identyfikowane w badaniu przesiewowym. Z czasem, gdy dieta niemowlęcia jest rozszerzana o inne pokarmy, wynik badania profilu acylokarnityn się normalizuje. W ten sposób badania przesiewowe noworodków metodą MS/MS pozwalają na wykrycie wrodzonej wady metabolizmu nie tylko u noworodków, ale czasem u ich dorosłych matek (materiał własny). 10, 12 Są one mimo częstego braku danych w wywiadzie o niepokojących objawach (przebieg bez- lub skąpoobjawowy) zagrożone ujawnieniem klinicznym deficytu MCC w sytuacjach związanych ze wzmożonym katabolizmem (operacja, ciężka infekcja, przedłużone głodzenie). U niektórych z nich stwierdzano wcześniej stany dekompensacji metabolicznej związane z infekcjami (materiał własny). 10
Przeanalizowano przebieg choroby w grupie 14 pacjentów zidentyfikowanych w badaniach przesiewowych noworodków (w wieku 1,75-6,5 roku), jak również ich czworga rodzeństwa (w wieku 4,5-12,25 roku) oraz 7 matek (w wieku 22-38 lat) zdiagnozowanych w trakcie poszerzonych badań rodzinnych. 13 Wszyscy byli bez objawów poza jednym dzieckiem z opóźnionym rozwojem mowy i 2 z wywiadem nietolerancji wysiłku fizycznego i osłabieniem kończyn. Autorzy dokonali analizy 37 przypadków opublikowanych wcześniej, z której wynikało, że 27% przypadków było bezobjawowych, 11% dzieci zmarło, 32% miało opóźniony rozwój psychoruchowy, 16% drgawki i 49% niespecyficzne objawy neurologiczne. Na podstawie powyższych danych wysunięto sugestię, że być może nie wszystkie objawy mogły być związane z deficytem MCC, a zwiększone stężenie C5OH w badaniu przesiewowym może nie mieć znaczenia klinicznego. Przeciwną opinię wyrażono w późniejszym opracowaniu Grünert i wsp. 10 Dokonano analizy danych klinicznych, biochemicznych, enzymatycznych i wyników badań molekularnych w grupie 88 pacjentów z rozpoznanym deficytem MCC, w tym 53 (60%) zidentyfikowanych w badaniach przesiewowych noworodków, 26 (30%) zdiagnozowanych wobec wystąpienia objawów klinicznych lub w ramach badań rodzinnych oraz 9 (10%) matek (w wieku 24-38 lat) zidentyfikowanych w trakcie diagnostyki różnicowej nieprawidłowości stwierdzanych u noworodków. Zanotowano trzy zgony. Pierwszy z powodu nagłego zatrzymania akcji serca w 33 dniu życia u dziecka zidentyfikowanego w ramach badań przesiewowych noworodków, z ciężką dekompensacją metaboliczną od pierwszego dnia życia, hipoglikemią, hiperamonemią i kwasicą mleczanową. Drugi zgon nastąpił u 6-tygodniowego niemowlęcia z uogólnioną wiotkością, drgawkami mioklonicznymi i ostrą dekompensacją metaboliczną. Trzeci zgon dotyczył 8-letniej dziewczynki, u której jednak objawy ze strony serca (tachykardia komorowa i nagłe zatrzymanie akcji serca) wydają się raczej spowodowane wykrytymi mutacjami w genie dla receptora rjanodyny RyR2, odpowiadającymi za nieprawidłowy napływ wapnia w komórkach mięśnia sercowego, tj. chorobą niezwiązaną przyczynowo z deficytem MCC. U 34 z 80 pacjentów (tj. w ponad 40%; w 8 przypadkach brakuje danych) występowały objawy kliniczne, od ciężkich dekompensacji metabolicznych (z encefalopatią, ketokwasicą i hipoglikemią) do objawów mięśniowych, upośledzonego rozwoju, deficytu uwagi i nadpobudliwości. W grupie 53 dzieci zdiagnozowanych w ramach badań przesiewowych noworodków 36 nie miało objawów, a u 13 stwierdzano różne objawy kliniczne (w czterech przypadkach brakuje danych). Oznacza to, że prawie co czwarte dziecko zidentyfikowane w fazie przedobjawowej ujawniło objawy kliniczne deficytu MCC. W tym w pięciu przypadkach wystąpiły pełnoobjawowe incydenty dekompensacji metabolicznej. Co ciekawe, w przypadkach wykrytego deficytu MCC pochodzenia odmatczynego u jednej z matek występowało przewlekłe zmęczenie, a poza tym nie stwierdzano żadnych niepokojących objawów, u drugiej jednak występowały nawracające dekompensacje metaboliczne w przebiegu infekcji gorączkowych, a także kardiomiopatia, parestezje i epizod udaru mózgu.
Własne wyniki analizy przypadków rozpoznanych w kraju wykazały dużą wykrywalność deficytu MCC w ramach badań przesiewowych noworodków, tj. metodą MS/MS. 14 Deficyt MCC rozpoznano ogółem u 42 osób, w tym na podstawie skriningu selektywnego wykryto 11 przypadków, diagnostyki rodzinnej – 5 przypadków, zaś w badaniach przesiewowych noworodków zidentyfikowano 19 dzieci i 7 matek. Poza grupą pacjentów zdiagnozowanych w ramach skriningu selektywnego i jedną z matek (u której w wywiadzie był przebyty w wieku 9 lat zespół Reye’a o nieustalonej wówczas etiologii) nie obserwowano niepokojących objawów. Najdłuższy okres obserwacji pacjentów zidentyfikowanych w przesiewowych badaniach noworodków wynosił jednak tylko 10 lat, a najpóźniejszy wiek ujawnienia się deficytu MCC w analizowanej grupie pacjentów z objawami to 12 lat.
Wśród 11 pacjentów z objawami zwracały uwagę: wymioty (u 4), drgawki lub ich ekwiwalenty (u 5), opóźnienie rozwoju psychoruchowego (u 3) i inne objawy neurologiczne. Hipoglikemię stwierdzono tylko u jednego pacjenta.
Stężenie C5OH-karnityny w suchej kropli krwi w momencie rozpoznania wynosiło 0,72-41,0 μmol/l u pacjentów z objawami i 0,51-31,5 μmol/l (wartości referencyjne <0,47 μmol/l) u pacjentów poddanych badaniom przesiewowym. Znaczący wzrost C5OH w suchej kropli krwi (>3,00 umol/l) stwierdzono odpowiednio w 45 i 48% przypadków zdiagnozowanych w skriningu selektywnym i przesiewowym badaniu noworodkowym. W chwili rozpoznania, niezależnie od sposobu diagnostyki, wydalanie 3-HIVA i MCG następowało u wszystkich pacjentów z wyjątkiem jednego, u którego pozostawało niewykrywalne przez 11 kolejnych miesięcy dalszej obserwacji. Masywne zwiększenie stężenia 3HIVA i MCG, definiowane jako większe niż 500 mmol/mol kreatyniny i 200 mmol/mol kreatyniny (odpowiednio wartości referencyjne <20 i niewykrywalne), było stwierdzane u około połowy pacjentów diagnozowanych zarówno w diagnostyce objawowej, jak i przedobjawowej.
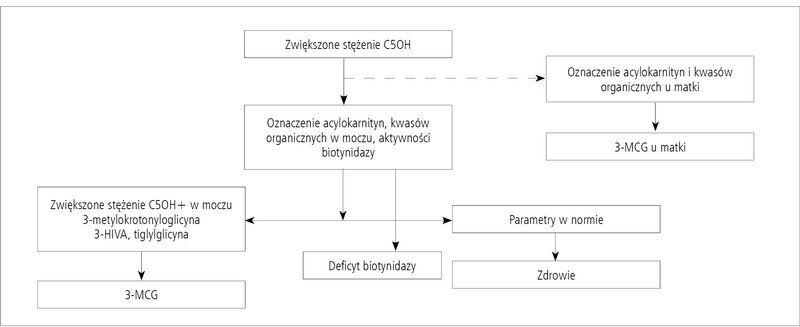
Rycina 2. Schemat postępowania w przypadku zwiększonego stężenia C5OH w MS/MS.
Wobec skomplikowanego procesu diagnostycznego w przypadku deficytu MCC autorzy proponują korzystanie z algorytmu diagnostycznego (ryc. 2), który powinien być pomocny w procesie różnicowania ewentualnych przyczyn zwiększonego stężenia C5OH u noworodka.

Tabela. Pierwszoplanowe badania w przypadku zwiększonego stężenia C5OH w badaniu przesiewowym noworodków
Deficyt biotynidazy powinien być wykluczony już na pierwszym etapie diagnostyki różnicowej. Jest on wrodzoną wadą metabolizmu dziedziczącą się w sposób autosomalny recesywny, przebiegającą z postępującymi objawami neurologicznymi (w tym drgawki oporne na leczenie, hipotonia, ataksja) i skórnymi (wysypki, wyłysienie) oraz zaburzeniami biochemicznymi (kwasica mleczanowa). Może się ujawniać już tuż po urodzeniu i wówczas manifestuje się najczęściej śpiączką (obraz typowy dla deficytu syntetazy holokarboksylaz). W pierwotnym deficycie biotynidazy choroba ujawnia się w okresie niemowlęcym, ale także może przebiegać skrycie. Skutecznym leczeniem jest stała, wcześnie wprowadzona suplementacja biotyny. Deficyt biotynidazy nie wymaga modyfikacji postępowania dietetycznego. U dzieci z objawami sugerującymi deficyt biotynidazy wskazane jest włączenie biotyny (w dawce 15-20 mg/24 h) jeszcze przed uzyskaniem wyników weryfikujących to rozpoznanie, a odstawienie w przypadku wykluczenia deficytu.
Zwiększone stężenie C5OH u noworodka wymaga wykonania profilu kwasów organicznych w moczu, tzn. przeprowadzenia diagnostyki różnicowej w kierunku następujących acydurii organicznych:
- deficytu liazy 3-hydroksy-3-metyloglutarylo-koenzymu A
- deficytu beta-ketotiolazy
- acydurii 2-metylo-3-hydroksymasłowej
- acydurii 3-metyloglutakonowej
Wobec wykazania obecności metabolitów typowych dla deficytu MCC u matki wskazane jest objęcie jej opieką ośrodka medycyny metabolicznej, w tym przeprowadzenie pełnej diagnostyki u niej i u dziecka.
Utrzymywanie się zwiększonego stężenia C5OH i wykazanie wydalania z moczem 3-HIVA i MCG (zwłaszcza przy prawidłowych wynikach tych badań u matki) wskazują na rozpoznanie deficytu MCC u dziecka. W celu ostatecznego potwierdzenia choroby wskazane byłoby przeprowadzenie badań enzymatycznych w fibroblastach skóry lub limfoblastach (niedostępnych w kraju) i/lub molekularnych (sekwencjonowanie genów MCCC1 i MCCC2). Po rozpoznaniu deficytu MCC dziecko powinno pozostać pod opieką i kontrolą kliniczno-dietetyczną ośrodka medycyny metabolicznej, wskazane jest też udzielenie pisemnej porady genetycznej rodzinie.
Postępowanie terapeutyczne w deficycie MCC
Zalecenia terapeutyczne w deficycie MCC zależą od ciężkości obrazu klinicznego i określenia ryzyka progresji choroby. Rozpoczęcie leczenia dietetycznego u pacjentów z rozpoznanym objawowo deficytem MCC nie budzi wątpliwości. Podstawową modyfikacją żywienia jest ograniczenie spożycia leucyny, pozwalające na zminimalizowanie powstawania nieprawidłowych acylokarnityn, przy równoczesnym utrzymaniu dodatniego bilansu azotowego, warunkującego prawidłowy rozwój organizmu. 15 W diecie noworodka/niemowlęcia zmniejszenie podaży leucyny jest możliwe dzięki zastąpieniu części mleka modyfikowanego specjalnym preparatem białkozastępczym, niezawierającym tego aminokwasu. Podobnie jest przy karmieniu piersią, które wymaga ograniczenia dobowej ilości pokarmu w zależności od stanu klinicznego i wyników biochemicznych. Do końca pierwszego roku życia rozszerzanie diety o pokarmy stałe powinno być zgodne z przyjętym schematem żywienia niemowląt. Wyjątek stanowią produkty bogatobiałkowe, takie jak mięso, żółtko, twaróg, które mogą być całkowicie wykluczone z jadłospisu dziecka lub być dozwolone (najczęściej zamiennie) w bardzo ograniczonych i ściśle określonych ilościach. W kolejnych latach życia pacjenci z deficytem MCC pozostający na diecie z ograniczeniem leucyny wymagają regularnego monitorowania tempa rozwoju i wszystkich parametrów stanu odżywienia. Przy zbyt restrykcyjnym ograniczeniu leucyny (tego egzogennego aminokwasu) istnieje, podobnie jak to obserwowano u dzieci z fenyloketonurią, ryzyko spowolnienia rozwoju fizycznego, szczególnie w pierwszych 3-4 latach życia, co jednak nie wydaje się tak groźne jak niekorzystny wpływ takiej sytuacji na rozwój intelektualny małego dziecka. 16, 17 Ze względu na małe zapasy energetyczne u niemowląt/dzieci stany katabolizmu wywołane infekcją z gorączką, wymiotami lub biegunką mogą szybko doprowadzić do dekompensacji metabolicznej z jej konsekwencjami. Dlatego ważnym zaleceniem dla rodziców dziecka z deficytem MCC jest unikanie przedłużonego głodzenia zarówno w stanie zdrowia, jak i w czasie przejściowej choroby. Szczególne postępowanie w sytuacjach zagrożenia jest zalecane także wszystkim pacjentom bez objawów z deficytem MCC. Natomiast dieta z ograniczeniem leucyny lub białka naturalnego raczej nie jest zalecana dla niemowląt/dzieci bez objawów z tą wrodzoną wadą metabolizmu (zgodność opinii zespołu ekspertów oceniona metodą Delphi - 85%). 1
W dostępnym piśmiennictwie nie opublikowano dotychczas randomizowanych badań oceniających wpływ diety o obniżonej zawartości leucyny na brak lub opóźnienie wystąpienia objawów klinicznych. Z drugiej strony opisywane przypadki dzieci z deficytem MCC bez objawów, u których wystąpiła kryza metaboliczna na skutek odstąpienia od zalecanego leczenia i właściwego postępowania, wskazują na stałe zagrożenie ujawnienia się pełnego obrazu choroby, wymagające zdecydowanej zmiany zaleceń terapeutycznych. Przedstawiono na przykład przypadek nagłej senności ze znaczną kwasicą metaboliczną i hipoglikemią w przebiegu infekcji górnych dróg oddechowych u 19-miesięcznej dziewczynki, zdiagnozowanej w badaniach przesiewowych, u której rodzice zaprzestali stosowania diety z ograniczeniem białka naturalnego, a także suplementacji L-karnityny. 18
Zwykle zaleca się u pacjentów z deficytem MCC podaż L-karnityny, która ma działanie odtruwające, ponieważ wiąże w organizmie toksyczne metabolity, tworząc izowalerylokarnitynę wydalaną z moczem. Dawkowanie zależy od aktualnego stężenia wolnej karnityny w surowicy, które należy regularnie monitorować. Najczęściej stosowana dawka L-karnityny to ok. 50 mg/kg/24 h. Często w chwili rozpoznania u pacjentów z objawami wykazywany jest wtórny niedobór wolnej karnityny w surowicy, podczas gdy stężenie karnityny całkowitej pozostaje w normie.
W stanach zagrażających wystąpieniem dekompensacji metabolicznej – najczęściej są to infekcje (zwłaszcza gorączkowe), przedłużone głodzenie, szczepienia, urazy lub nadmierna podaż białka – bezwzględnie należy zapewnić podaż kalorii przez częste pojenie słodkimi napojami lub polimerami glukozy i ograniczyć podaż białka naturalnego w diecie. Gdy jest to niemożliwe (np. z powodu braku łaknienia i/lub wymiotów) i/lub rozwijają się niepokojące objawy (np. senność, drżenia, zaburzenia równowagi), niezbędna jest pilna hospitalizacja w celu przerwania stanu katabolizmu. Uzyskuje się to dzięki wlewom dożylnym stężonej glukozy (ok. 10 mg/kg/min), odstawieniu białka naturalnego (ale nie dłużej niż na 48 godzin) i stosowaniu L-karnityny doustnie lub dożylnie w dużych dawkach (ok. 100 mg/kg/24 h).
Podsumowanie
Deficyt MCC jako najczęściej identyfikowana choroba w badaniu przesiewowym noworodków metodą MS/MS powinien być znany pediatrom, którzy często (obok neonatologów) przeprowadzają diagnostykę różnicową i potem sprawują opiekę nad pacjentami wraz z monitorowaniem przebiegu choroby. Decydują również o leczeniu w stanie nagłym, kiedy znajomość właściwego postępowania terapeutycznego jest bardzo istotna i nieraz warunkuje rokowanie u dziecka.
Możliwość wystąpienia różnych fenotypów choroby i brak biomarkerów świadczących o spodziewanym przebiegu klinicznym (ani biochemiczne odchylenia w deficycie MCC, ani genotyp) nie pozwalają na przewidywanie obrazu klinicznego choroby ani decyzję, kogo leczyć. Klinicysta musi kierować się własnym doświadczeniem i odpowiednimi protokołami postępowania. 1 Czujność i indywidualne podejście do każdego pacjenta wydają się przy obecnym stanie wiedzy najważniejsze, a współpraca ze specjalistą pediatrii metabolicznej niezbędna.
Pacjenci z deficytem MCC zwykle nie mają żadnych objawów klinicznych w stanie dobrego zdrowia, ale może się u nich rozwinąć ciężka kwasica ketonowa z hipoglikemią podczas ostrego stresu, jakim jest infekcja. 19 Uważa się, że także pacjenci bez objawów z deficytem MCC wykrytym w ramach skriningu noworodkowego są zagrożeni wystąpieniem dekompensacji metabolicznej w sytuacji stresowej, o ile odpowiednie postępowanie profilaktyczne i leczenie nie będą włączone podczas ostrej infekcji. 18
Konflikt interesów
Autorzy nie zgłaszają żadnych konfiktów interesów związanych z treścią artykułu.

 Dodaj do ulubionych
Dodaj do ulubionych
