Onkologia
Zespół hipereozynofilowy i eozynofilia klonalna: wstępny algorytm diagnostyki w miejscu udzielania pomocy i aktualne metody leczenia
Ayalew Tefferi, MD1, Jason Gotlib, MD2, Animesh Pardanani, MBBS, PhD1
W skrócie
Nabytą eozynofilię można do celów praktycznych podzielić na eozynofilię wtórną, klonalną oraz samoistną. Do przyczyn wtórnej eozynofilii należą zakażenia pasożytnicze, choroby alergiczne, zapalenia naczyń, odczyny polekowe oraz chłoniaki. Eozynofilię klonalną odróżnia od eozynofilii samoistnej obecność histologicznych, cytogenetycznych lub molekularnych cech pierwotnego nowotworu szpiku. W klasyfikacji nowotworów hematologicznych wg WHO wyróżnia się dwie podkategorie eozynofilii klonalnej: przewlekła białaczka eozynofilowa (inaczej nieokreślona) oraz nowotwory mieloidalne/limfoidalne z obecnością eozynofilii i mutacji genów receptora płytkopochodnego czynnika wzrostu (PDGFR – platelet-derived growth factor receptor) α/β lub receptora dla czynnika wzrostu fibroblastów 1 (FGFR1 – fibroblast growth factor receptor 1). Eozynofilia klonalna może również towarzyszyć innym chorobom rozrostowym układu krwiotwórczego określonym w klasyfikacji WHO, takim jak przewlekła białaczka szpikowa, zespoły mielodysplastyczne, przewlekła białaczka mielomonocytowa i układowa mastocytoza. Kryteria rozpoznawcze zespołu hipereozynofilowego, który jest podkategorią eozynofilii samoistnej, to eozynofilia we krwi obwodowej rzędu 1,5 × 103/μl lub więcej utrzymująca się przez co najmniej 6 miesięcy (lub krócej jeżeli występują objawy wymagające stosowania leków zmniejszających liczbę eozynofilów), wykluczenie eozynofilii wtórnej i klonalnej, oznaki zajęcia narządów wewnętrznych oraz niewykrycie limfocytów T o nieprawidłowych fenotypach lub klonalnego rozrostu limfocytów. Niespełnienie tego ostatniego kryterium jest równoznaczne z rozpoznaniem wariantu limfocytowego hipereozynofilii, którą należy zaliczyć do eozynofilii wtórnych. W niniejszej pracy przedstawiamy uproszczony algorytm pozwalający na rozróżnienie różnorodnych przyczyn eozynofilii klonalnej i samoistnej oraz omawiamy obecne metody leczenia, w tym nowe leki (imatynib, alemtuzumab i mepolizumab).
Zwiększenie liczby granulocytów kwasochłonnych (eozynofilię) obserwuje się stosunkowo często na obszarach zwrotnikowych ipodzwrotnikowych, gdzie przyczyną tego stanu są przede wszystkim robaczyce przebiegające zzajęciem tkanek.1 Wkrajach Zachodu główne przyczyny eozynofilii wtórnej to choroby alergiczne lub zapalenia naczyń, odczyny polekowe oraz nowotwory niemieloidalne, choć zawsze należy rozważyć możliwość infestacji pasożytniczej, zwłaszcza uosób powracających zpodróży iimigrantów lub uchodźców niedawno przybyłych zregionów endemicznych.2 Odczyny polekowe przebiegające zeozynofilią iobjawami układowymi to groźne dla życia powikłania leczenia allopurynolem, karbamazepiną iinnymi lekami, wtym także niektórymi antybiotykami.3 Spośród chorób alergicznych izapaleń naczyń przebiegających zeozynofilią wtórną należy wymienić choroby płuc przebiegające ze zwiększeniem liczby eozynofilów, takie jak ostre iprzewlekłe eozynofilowe zapalenie płuc, alergiczna aspergiloza oskrzelowo-płucna oraz alergiczne ziarniniakowe zapalenie naczyń (zespół Churga-Strauss, przebiegający zeozynofilią, astmą, układowym zapaleniem naczyń iobecnością nacieków wpłucach).4 Eozynofilowe zapalenie żołądka ijelit może nie objawiać się eozynofilią we krwi; uważa się, że jest to choroba oodmiennej patogenezie wymagająca odrębnego leczenia.5,6
Wykluczenie eozynofilii wtórnej wymaga z reguły zebrania dokładnego wywiadu dotyczącego odbywanych podróży i stosowanych leków, wykonania badania przedmiotowego i badań laboratoryjnych, w tym zdjęcia RTG klatki piersiowej, powtarzanych badań w celu wykrycia jaj i pasożytów w kale (np. tęgoryjca dwunastnicy [Ancylostoma duodenale], przywr z rodzaju Schistosoma) i badań serologicznych w kierunku obecności podejrzanych pasożytów (np. Strongyloides stercoralis, Schistosoma spp., Toxocara spp., Filaria spp.).7,8 Różnicowane między eozynofilią samoistną przebiegającą z zajęciem narządów wewnętrznych oraz eozynofilią w przebiegu układowych zapaleń naczyń lub eozynofilowego zapalenia żołądka i jelit może jednak sprawiać trudności; w niektórych przypadkach możemy mieć do czynienia z różnymi postaciami spektrum klinicznego tej samej choroby. Jeżeli nie można szybko ustalić przyczyny eozynofilii wtórnej, rozsądnie jest przyjąć wstępne rozpoznanie eozynofilii klonalnej lub samoistnej i prowadzić dalszą celowaną diagnostykę w poszukiwaniu konkretnej jednostki chorobowej.
Podział eozynofilii klonalnej i samoistnej
Eozynofilia klonalna to rozrost nowotworowy eozynofilów będący częścią obrazu klinicznego pierwotnej choroby rozrostowej komórek linii mieloidalnej. Eozynofilia klonalna może zatem towarzyszyć każdemu procesowi rozrostowemu komórek mieloidalnych zdefiniowanemu w klasyfikacji nowotworów układu krwiotwórczego wg WHO (tabela).9 Klasyfikacja ta obejmuje dwie podkategorie eozynofilii klonalnej: przewlekłą białaczkę eozynofilową inaczej nieokreśloną (CEL-NOS – chronic eosinophilic leukemia, not otherwise specified) oraz nowotwory mieloidalne /limfoidalne z obecnością eozynofilii i mutacji receptora płytkopochodnego czynnika wzrostu (PDGFR) α/β lub receptora dla czynnika wzrostu fibroblastów 1.
Rozpoznanie eozynofilii samoistnej oznacza, że wykluczono eozynofilię wtórną oraz klonalną; u dzieci należy brać pod uwagę rzadko występującą eozynofilię wrodzoną. Zespół hipereozynofilowy (HES – hypereosinophilic syndrome) to podkategoria eozynofilii samoistnej. Jego rozpoznanie wymaga stwierdzenia eozynofilii rzędu 1,5 × 103/μl lub więcej we krwi obwodowej oraz uszkodzenia narządów wewnętrznych wywołanego przez eozynofile. Należy odróżnić zespół hipereozynofilowy od pojęcia hipereozynofilii, które oznacza po prostu obecność we krwi obwodowej granulocytów kwasochłonnych w liczbie ≥1,5 × 103/μl. Eozynofilię związaną z obecnością limfocytów o nieprawidłowym fenotypie lub z rozrostem klonalnym limfocytów lepiej nazywać hipereozynofilią limfocytową niż limfocytowym HES.
Rozróżnienie eozynofilii klonalnej i samoistnej ma charakter umowny. Doświadczenie kliniczne wskazuje, że w niektórych przypadkach HES jest w rzeczywistości objawem pierwotnej choroby rozrostowej szpiku. U niektórych pacjentów z HES wykazano na przykład monoklonalną proliferację eozynofilów,10,11 a u innych przejście choroby w nowotwór mieloidalny ujęty w klasyfikacji WHO.12-15 Ponadto u osób z eozynofilią klonalną i obecnością genu fuzyjnego FIP1L1-PDGFRA w okresie przed odkryciem tej mutacji, tj. przed 2003 r., często rozpoznawano właśnie HES.16
Algorytm diagnostyczny w przypadku podejrzenia eozynofilii klonalnej lub samoistnej
Diagnostyka różnicowa pacjenta z eozynofilią, która prawdopodobnie nie ma charakteru wtórnego, powinna uwzględniać pięć następujących możliwości: (1) nowotwory mieloidalne lub limfoidalne przebiegające z eozynofilią, związane z rearanżacją w obrębie genu PDGFR lub FGR1, (2) eozynofilia klonalna w przebiegu innego nowotworu mieloidalnego ujętego w klasyfikacji WHO, (3) CEL-NOS, (4) limfocytowa hipereozynofilia oraz (5) eozynofilia samoistna, w tym HES. Ostateczne rozpoznanie ustala się w kilkuetapowym procesie (ryc.), obejmującym staranną ocenę rozmazu krwi obwodowej i morfologii szpiku kostnego, analizę cytogenetyczną, badania molekularne, w tym badania przesiewowe w kierunku wykrycia mutacji FIP1L1-PDGFRA oraz fenotypowanie limfocytów krwi obwodowej i ocenę rearanżacji genów receptorów limfocytów T.
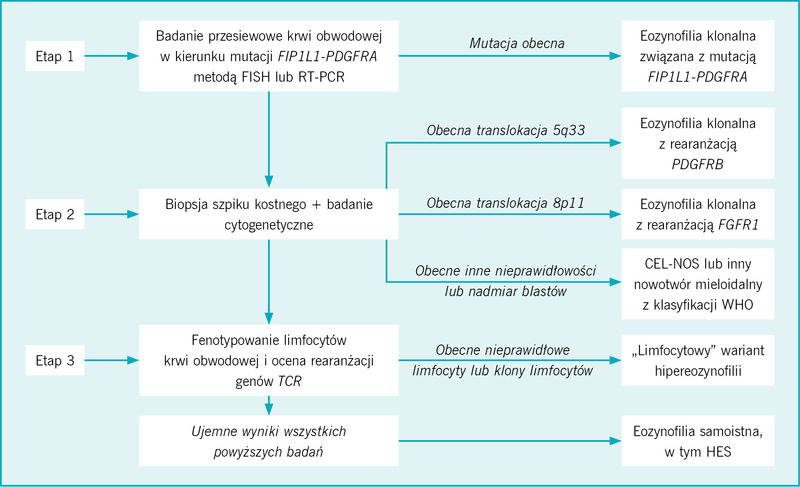
Rycina. Algorytm diagnostyki eozynofilii klonalnej lub samoistnej
Po analizie rozmazu krwi obwodowej i wyników badań laboratoryjnych w poszukiwaniu wskazówek sugerujących pierwotny nowotwór mieloidalny (np. obecność blastów we krwi obwodowej, obecność komórek dysplastycznych, monocytoza, podwyższona aktywność tryptazy w surowicy), których stwierdzenie jest bezwzględnym wskazaniem do niezwłocznego pobrania szpiku kostnego w celu ustalenia konkretnego rozpoznania, uzasadnione jest wykonanie badania przesiewowego w kierunku mutacji FIP1L1-PDGFRA we krwi obwodowej metodą hybrydyzacji fluorescencyjnej in situ (FISH – fluorescence in situ hybridization) lub łańcuchowej reakcji polimerazy z odwrotną transkryptazą (RT-PCR – reverse transcription polymerase chain reaction) (ryc.). Jeżeli uda się wykryć powyższą mutację, można odstąpić od badania szpiku kostnego, przyjąć robocze rozpoznanie nowotworu mieloidalnego z mutacją FIP1L1-PDGFRA i rozpocząć leczenie imatynibem (leczenie omówiono w następnym podrozdziale). W naszej praktyce preferujemy jednak wykonanie badania szpiku kostnego w celu wykluczenia obecności istotnych rokowniczo morfologicznych lub cytogenetycznych markerów ewolucji klonalnej.
Jeżeli badanie przesiewowe krwi obwodowej w kierunku mutacji FIP1L1-PDGFRA da wynik ujemny, kolejnym etapem jest wykonanie badania szpiku kostnego i badań cytogenetycznych w celu wykrycia innych nieprawidłowości wskazujących na klonalną ekspansję eozynofilów. Należy po pierwsze zwrócić uwagę na translokacje 5q33, 4q12 czy 8p11.2, których obecność wskazuje, że eozynofilia klonalna jest związana z rearanżacją w obrębie odpowiednio PDGFRB, PDGFRA oraz FGFR1 (ryc.). Ten etap jest niezwykle ważny dla planowania postępowania terapeutycznego, ponieważ wykrycie translokacji 5q33 lub 4q12 wskazuje na możliwość dobrej odpowiedzi na leczenie imatynibem, podczas gdy translokacje 8p11.2 stwierdza się u chorych z agresywnymi nowotworami mieloidalnymi opornymi na obecnie stosowane schematy farmakoterapii (leczenie omówiono w następnym podrozdziale). W przypadku wykrycia translokacji 5q33 lub 8p11.2 należy ponadto wykonać badanie metodą FISH lub RT-PCR w celu potwierdzenia nieprawidłowości odpowiednio PDGFRB lub FGFR1.
Ocena morfologii komórek szpiku kostnego pomaga także wykluczyć inne możliwe nowotwory mieloproliferacyjne ujęte w klasyfikacji WHO (tab.).
|
Tabela. Klasyfikacja nowotworów mieloidalnych |
|
Ostra białaczka szpikowa i powiązane choroby |
|
Nowotwory mieloproliferacyjne |
|
Przewlekła białaczka szpikowa z obecnością BCR-ABL1 |
|
Przewlekła białaczka neutrofilowa |
|
Czerwienica prawdziwa |
|
Pierwotne zwłóknienie szpiku (mielofibroza) |
|
Nadpłytkowość samoistna |
|
Przewlekła białaczka eozynofilowa inaczej nieokreślona |
|
Mastocytoza |
|
Nowotwory mieloproliferacyjne niesklasyfikowane |
|
Zespoły mielodysplastyczne |
|
Cytopenia oporna na leczenie z dysplazją jednej linii komórkowej |
|
|
|
|
Niedokrwistość oporna na leczenie z pierścieniowatymi syderoblastami |
|
Cytopenia oporna na leczenie z dysplazją wielu linii komórkowych |
|
Niedokrwistość oporna na leczenie z nadmiarem blastów |
|
|
|
Zespół mielodysplastyczny z izolowaną delecją del(5q) |
|
Zespół mielodysplastyczny niesklasyfikowany |
|
Zespoły mielodysplastyczne/nowotwory mieloproliferacyjne |
|
Przewlekła białaczka mielomonocytowa |
|
Atypowa przewlekła białaczka szpikowa bez rearanżacji BCR-ABL1 |
|
Młodzieńcza postać białaczki mielomonocytowej |
|
Zespoły mielodysplastyczne/nowotwory mieloproliferacyjne niesklasyfikowane |
|
|
Nowotwory mieloidalne i limfoidalne przebiegające z eozynofilią i nieprawidłowościami genetycznymi |
|
Nowotwory mieloproliferacyjne przebiegające z eozynofilią i rearanżacją PDGFRA |
|
Nowotwory mieloproliferacyjne przebiegające z eozynofilią i rearanżacją PDGFRB |
|
Nowotwory limfo- i mieloproliferacyjne przebiegające z eozynofilią i rearanżacją FGFR1 |
Szczególne znaczenie w diagnostyce różnicowej ma wykluczenie układowej mastocytozy, przewlekłej białaczki mielomonocytowej oraz CEL-NOS.17 Rozpoznanie układowej mastocytozy wymaga stwierdzenia obecności skupisk komórek tucznych o nieprawidłowej morfologii, wykazania nieprawidłowej ekspresji CD25 na komórkach tucznych lub obecności mutacji KITD816V.18 Rozpoznanie przewlekłej białaczki mielomonocytowej wymaga stwierdzenia obecności ponad 1 × 103/μl monocytów we krwi obwodowej. Rozpoznanie CEL-NOS należy brać pod uwagę, jeżeli liczba eozynofilów we krwi obwodowej wynosi co najmniej 1,5 × 103/μl oraz stwierdza się cytogenetyczne lub morfologiczne cechy nowotworu mieloidalnego o inaczej nieokreślonym charakterze. CEL-NOS różnicuje się z HES na podstawie stwierdzenia nieprawidłowości w badaniu cytogenetycznym lub wykazania ponad 2% blastów we krwi obwodowej lub ponad 5% komórek blastycznych w szpiku kostnym.19
Rozpoznanie eozynofilii samoistnej, w tym HES jako podkategorii, wymaga wykluczenia eozynofilii wtórnej i klonalnej oraz niestwierdzenia obecności limfocytów T o nieprawidłowym fenotypie lub rozrostu klonalnego limfocytów T. Rozpoznanie HES (jako podkategorii eozynofilii samoistnej) wymaga dodatkowo stwierdzenia eozynofilii we krwi obwodowej ≥1,5 × 103/μl utrzymującej się przez co najmniej 6 miesięcy (lub krócej, jeżeli występują objawy wymagające stosowania leków zmniejszających liczbę eozynofilów) i cech zajęcia narządów wewnętrznych. U wszystkich chorych z podejrzeniem HES należy określić fenotyp limfocytów krwi obwodowej i wykonać badanie rearanżacji genów dla receptorów limfocytów T w celu wykluczenia limfocytowego wariantu hipereozynofilii, rozpoznawanego na podstawie stwierdzenia rozrostu klonalnego limfocytów T lub obecności limfocytów T o nieprawidłowym fenotypie (np. CD3–CD4+).20